Synaptic Plasticity Project
研究内容の紹介
自閉症は近年増加傾向にあり、発症機序を含め、治療法は確立されていません。また、知的障害については医療の対象になっておらず、教育や福祉、介護的な支援が行われているだけです。本プロジェクトでは、自閉症や知的障害、難治性てんかんなど、脳の発達の異常を研究対象とし、そのメカニズムを解析することによって、新しい治療法の開発を目指しています。
そのための戦略として、これらの病態に共通してみられるシナプス、特に樹状突起スパイン(興奮性シナプスの後部)の異常に注目しています(Epilepsia 15:55-80,1974; Brain Res Rev. 39:29-54, 2002、図1参照)。例えば、自閉症の原因遺伝子産物として、neuroligin-3/4やneurexin-1(接着分子とそのリガンド、Nat Genet. 34:27-9, 2003; Am J Hum Genet. 82:199-207, 2008)、shank3(PSDタンパク質、Nat Genet. 39:25-7, 2007)、FMRP(RNA結合タンパク質、Trends Neurosci. 27:370-7, 2004)、Ube3A(ユビキチンリガーゼ、Mol Psychiatry 4:64-7, 1999)などシナプスに関わるタンパク質が多数報告されており、それらの変異によってシナプスの形が大きく変化します(Proc Natl Acad Sci U S A. 94:5401-4, 1997; Hum Mol Genet. 13:1471-7, 2004; Nat Genet. 39:25-7, 2007; Hum Mol Genet. 17:111-8, 2008)。これらの事実は、シナプス形態の調節に関わるタンパク質の異常が自閉症や知的障害、てんかんなどの原因になり得ることを示しています。

図1.脳発達障害にみられるシナプス形態変化
A:正常発達
B:精神遅滞患者
C:精神遅滞患者(重度)
D:脆弱X症候群患者
(Fiala et al., Brain Res Rev. 39:29-54, 2002 より)
私たちは、シナプスの発達や神経活動に伴って増えるタンパク質(低分子量Gタンパク質Rheb、カドヘリンの一種arcadlin/pcdh8、神経突起の枝分かれを調節するneuritin、炎症物質プロスタグランジンの合成酵素 cyclooxygenese-2 (COX-2)とmicrosomal prostaglandin E synthetase (mPGES-1)、グルタミン酸受容体の細胞内移行を調節するarcなど)を発見しました。これらの分子を調べることによって、自閉症や知的障害、難治性てんかんなどの病態を明らかにし、それに基づいた新しい治療薬の探索を目指しています。以下に現在進めている研究内容を紹介します。
1)結節性硬化症の新しいメカニズムと治療薬
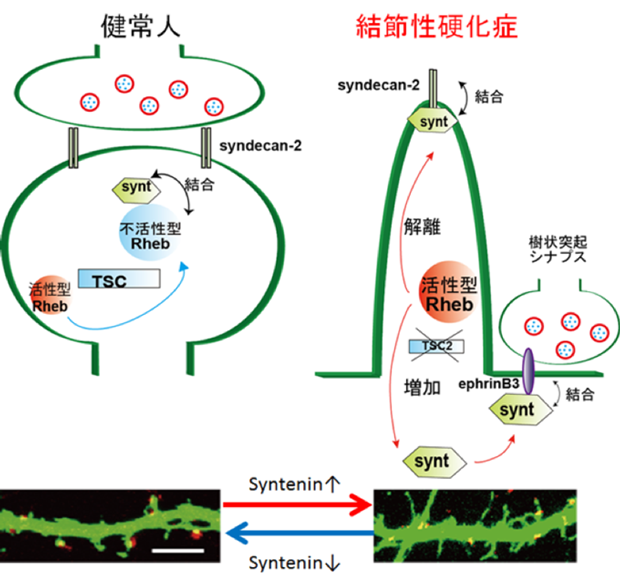
図2.結節性硬化症における樹状突起スパイン形成不全のメカニズム
結節性硬化症ではRhebが活性化する結果、Rhebと結合していたsynteninが離れ、syndecan-2と結合するため、スパイン形成が阻害されることがわかりました。Synteninを減少させると、結節性硬化症でも正常なシナプスに変わりました。
結節性硬化症は、全身に過誤腫とよばれる良性腫瘍ができる疾患ですが、臨床的に最も問題となるのは、乳児期から始まる難治性てんかんや知的障害です。さらに自閉症も高率(30-60%)に合併することが知られています。結節性硬化症の原因遺伝子はTsc1とTsc2ですが、これらは低分子量Gタンパク質Rhebを不活化(GDP結合型に)することが報告されました。すなわち、結節性硬化症患者ではTsc1あるいはTsc2の変異によって、Rhebが活性型(GTP結合型)となり、その下流のmTOR(ラパマイシンの標的分子)を活性化するため、病気を発症すると考えられてきました(図3B)。
しかし、mTORC1阻害薬のラパマイシンが、結節性硬化症のシナプス異常を改善しないこと(Yasuda, Sugiura et al., Sci. Rep. 2014)から、私たちはmTORC1ではなく、Rhebの活性化がシナプス異常の原因ではないかと考え、以下の新しいシナプス制御経路を発見しました。すなわち、
1)Rhebは正常ではsynteninというPDZ蛋白質と結合していること、
2)結節性硬化症ではRhebが活性化すると、synteninがRhebから離れること、
3)遊離したsynteninが、syndecan-2というシナプス形成因子と結合し、CASKという別のPDZ蛋白質(大田原症候群の原因遺伝子の一つ)を排除すること、です(Sugiura et al., Nat. Commun. 2015)。
この新しいメカニズムに基づけば、難治てんかんを起こす病気として別々に考えられてきた疾患が、実は同じメカニズムの異常であると理解できます。そして、結節性硬化症の中枢病態の治療には、ラパマイシンではなく、Rheb-synteninを標的とした薬物の使用がより有効であることが期待されます。さらに、Rhebそのものが他の難治てんかん発作によっても誘導・活性化される(Yamagata et al., J. Biol. Chem. 1994)ことから、Rheb-syntenin系はてんかんに伴う他の知的障害や自閉症の病態に関与している可能性も考えられます。私達の研究室では、これらを標的とした薬物の探索や開発を進めています。
2)てんかん発作による記憶障害のメカニズム
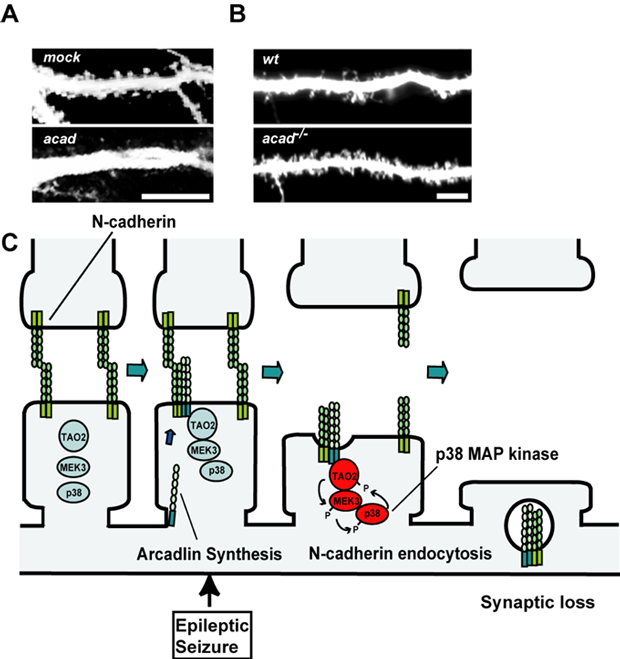
図3.神経活動依存的に発現するプロトカドヘリンarcadlinによるシナプス減少の分子メカニズム
A:正常ラットニューロンにarcadlinを過剰発現させるとシナプスが減少しました。
B:arcadlinノックアウトマウスではシナプスが増加していました。
C:神経活動によって誘導されたarcadlinはシナプスへ運ばれ、セリン-スレオニンキナーゼTAO2キナーゼを介してp38MAPキナーゼを活性化します。活性化したp38がTAO2をリン酸化することにより、arcadlin-N-cadherin複合体が内在化し、シナプス密度が減少します。
Arcadlin (pcdh8)はカドヘリンスーパーファミリーに属し、細胞同士の接着を司る分子の一つです。脳においてはクラシカルカドヘリンのN-cadherinが主として発現しており、シナプスの接着を維持しています。脳には、他にプロトカドヘリンと呼ばれる分子群が存在し、私達も神経活動により誘導されるarcadlin/pcdh8を見つけました(Yamagata et al., J. Biol. Chem. 1999)。これまでの研究により、1)arcadlinがシナプスにおいてN-cadherinと結合していること、2)arcadlinがホモ接合するとTAO2というリン酸化酵素が活性化され、MEK3-p38 MAPキナーゼ系が活性化されること、3)p38 MAPキナーゼがTAO2をリン酸化することによって、arcadlin/N-cadherin複合体のエンドサイトーシスが生じること、を明らかにしました(Yasuda, Tanaka, Sugiura et al., Neuron 2007; 図3)。すなわち、てんかん発作が起きるとN-cadherinがスパイン膜から内在化し、スパイン密度が減少する結果、記憶障害が生じると考えられます。このスパインの減少が、てんかん発作後の記憶障害の原因と考えられます。
さらに、TAO2キナーゼ遺伝子は染色体16p11.2領域に存在しますが、その微小欠失が自閉症、微小重複が統合失調症に関連することが報告されています(Weiss et al., N Engl J Med. 2008; McCarthy et al., Nat Genet. 2009)。TAO2はシナプス密度を制御することから、上記の病態に関与するかどうかも興味のあるところです。
3)側頭葉てんかん難治化のメカニズム
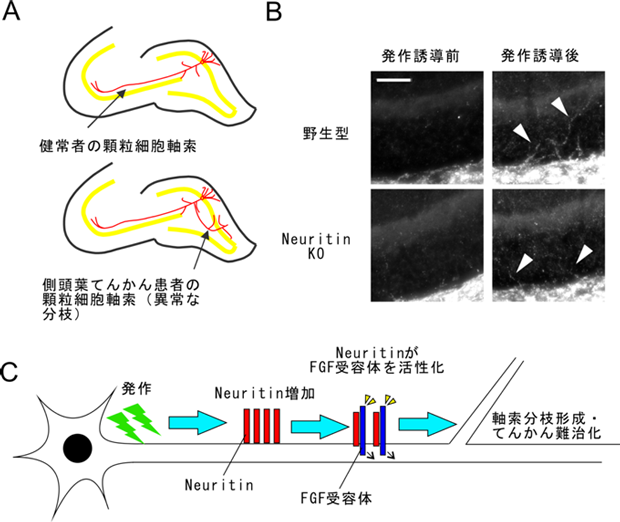
図4.Neuritinにより制御されるてんかん難治化のメカニズム
A:てんかん患者では顆粒細胞軸索に異常な分枝が形成されます
B:Neuritinノックアウトマウスでは発作誘導後に顆粒細胞軸索の分枝形成が観察されませんでした。
C:発作によりNeuritinの発現が上昇し、FGFシグナルが活性化することで、軸索分枝が形成されます。
苔状線維発芽とはてんかん患者、およびてんかんモデル動物で観察される現象です。海馬歯状回に存在する神経である顆粒細胞の軸索は通常は海馬CA3領域に投射しますが、てんかん発作により異常な軸索分枝が形成され、歯状回にも投射するようになり自己回路を作ります(図4A)。この現象は苔状線維発芽と呼ばれ、側頭葉てんかん難治化の原因の一つと考えられています(Houser et al., Epilepsia 2012)。
Neuritin (CPG15)は神経活動依存的に発現が上昇するタンパク質であり、発作誘導後海馬歯状回でも発現が上昇します(Naeve et al., Proc. Natl. Acad. Sci. 1997)。私達はneuritinが顆粒細胞の軸索分枝形成を誘導することを見出しました。また、neuritinを欠損するマウスではてんかんの発作の悪化速度が遅く、苔状線維発芽もあまり見られませんでした(Shimada et al., J. Neurosci. 2016)(図4B)。すなわち、てんかん発作によるneuritinの発現誘導が苔状線維発芽を引き起こし、てんかんの難治化につながると考えられました。また、neuritinはFGF受容体と結合し、軸索分枝が誘導されると考えらます(図4C)。実際に、FGFシグナルを阻害することでNeuritin依存的な軸索分枝形成を抑えることができました。これらの結果から、neuritin/FGFのシグナルを抑えることでてんかんの難治化を予防することができるのではないかと予想されます。
4)血管内皮細胞を介する神経細胞脱落のメカニズム
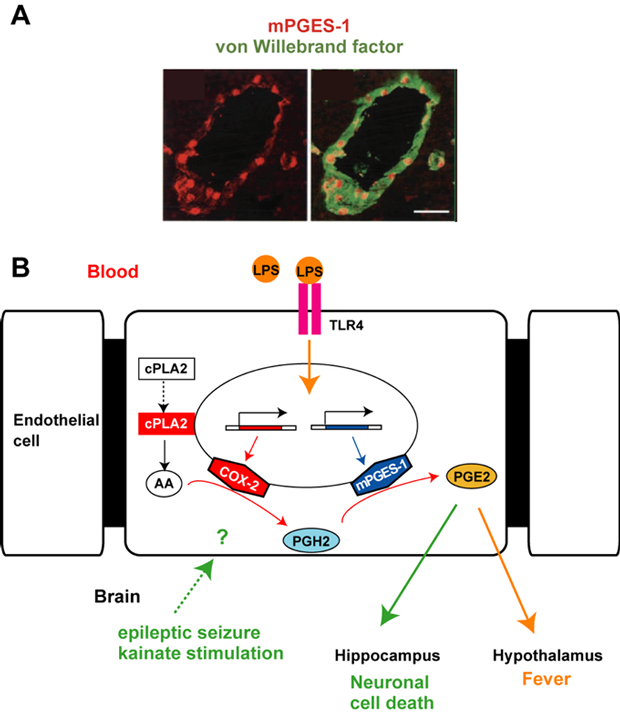
図5.血管内皮細胞に発現するCOX-2、mPGES-1による発熱、神経細胞死のメカニズム
A:リポポリサッカライド(LPS)投与により血管内皮に発現したmPGES-1(赤)と血管内皮細胞のマーカーであるvon Willebrand 因子(緑)との二重染色像
B:血管内皮に発現したCOX-2とmPGES-1により、プロスタグランジンE2(PGE2)が合成され、発熱や神経細胞死を制御する。
私達は、てんかん発作や脳虚血発作等によってプロスタグランジン合成酵素COX-2が神経細胞で一過性に誘導されることを初めて見出しました(Yamagata et al., Neuron 1993)。さらに、発熱を起こす細菌内毒素(LPS)やけいれん誘発薬(カイニン酸)投与によっても、COX-2と下流のPGE2合成酵素mPGES-1が血管内皮細胞において誘導されることを明らかにしています。COX-2とmPGES-1の誘導により、内皮細胞内でPGE2が合成され、視床下部の体温調節中枢に作用することにより発熱を(Yamagata et al., J. Neurosci. 2001)、またアストロサイトを介して海馬の神経細胞脱落を促進する(Takemiya et al., J. Neurosci. Res. 2010; Takemiya et al., Neurochem. Int. 2011)ことを明らかにしました(図5)。
最後に
このように、私たちは独自の観点から自閉症や知的障害・難治てんかんの分子・細胞メカニズムを明らかにしてきました。現在、それらに基づく治療薬の探索を進めており、数年以内に新しい治療薬の開発を目指しています。
これらの疾患研究は世界的にも競争が激しい分野ですが、私たちはこれまで独自の病態モデルを発表してきました。メカニズムが明らかになれば、薬の開発が可能になります。自閉症や知的障害は現在のところ有効な治療法がありません。新たに見出したメカニズムにもとづいて治療薬の探索を行い、臨床へ還元していきたいと願っています。