ホーム > 研究内容 > 松田チーム
研究内容 【松田チーム】
ミトコンドリアの品質管理に着目した、パーキンソン病発症メカニズムの研究
メンバー
| 参事研究員 | 松田 憲之 チームリーダー(兼プロジェクトリーダー) |
|---|---|
| 研究員 | 山野 晃史 |
| 小谷野 史香 | |
| ポスドク | 小島 和華 |
| 実験補助員 | 木村 まゆみ |
| 山岸 麗香 | |
| 研修生 | 霍 安妮(東京都立大・博士課程) |
| 宇田川 智里(お茶女・修士課程) | |
| 荻原 禅(北里大・学部生) | |
| 宿院 丈(北里大・学部生) |
研究内容の紹介
家族性パーキンソン病の責任遺伝子産物 PINK1 と Parkin の研究
- 1: 一般の方向けの説明
- 2: 研究系の学生や医師・専門家向けの説明
一般の方へ
以下は一般の方を対象に、私たちのチームの代表的な研究テーマのひとつ、“パーキンソン病の発症に関連した不良ミトコンドリア分解システム”について紹介するものです。
ユビキチンプロジェクト全体として現在取り組んでいる研究テーマについてより簡単に紹介する<一般の方向けプロジェクトページ>も是非ご覧ください。詳しい内容や成果は<研究系の学生や医師・専門家向け>のページをご覧下さい。
(1)パーキンソン病とは?
パーキンソン病は、脳の「黒質」と呼ばれる部分を構成する神経細胞が減ってしまうこと(神経細胞死)によって、様々な異常が発生する病気です。

代表的な運動症状として、
(a) 動作の開始に極端に時間がかかり、始めた動作も非常にゆっくりとしか行なうことができない。
(b) 安静時に手足や顎にふるえがみられる。
(c) 筋肉が緊張して、関節を受動的に屈伸するとガクガクとした抵抗を受ける。
(d) 姿勢を保持することが難しく、転倒しやすい。
(e) すり足、小股、前屈姿勢などで歩行がうまくできない。
などがあり、さらに非運動症状として、抑うつや幻覚、自律神経症状や睡眠障害がみられます。進行すると、自立した生活が困難になってしまう恐れがあります。
日本では1000人に1人以上の頻度で発症するといわれており、15万人を超える患者さんがいると推定されています。高齢化社会を迎える今日、予防法や治療法の確立が求められています。
多くの場合、老化に伴い50~60歳で発症する「孤発性」(遺伝的な要素がない)の病気ですが、少数ながら「家族性」(遺伝的な要因があり、家系内で発症する)の場合があります。私たちはこの「家族性」のパーキンソン病に注目して、研究を行なっています。
(2)なぜ「家族性」のパーキンソン病に注目するのか?
病気の原因を調べるためには様々な方法があります。最も代表的なものが、患者さんにどんな異常が起こっているのかを細かく診察したり、組織レベルで観察して、病気の原因を明らかにする方法です。
一方で、病気に関係のある遺伝子を見つけ、それらが細胞の中でどのような役割を担うのかを調べることで、その「故障」によって引き起こされる病気を理解しようとする分子遺伝学的方法があります。
私たちはそのような分子遺伝学的方法によって「なぜパーキンソン病になるのか」を分子レベルで理解するために、家族性パーキンソン病の原因遺伝子から作り出されるタンパク質:Parkin(パーキン) と PINK1(ピンク1) の研究を行っています。
原因遺伝子というのは、PINK1/Parkin が病気を引き起こすという意味ではなくて、PINK1/Parkin が異常になるとパーキンソン病が引き起されるという意味です。PINK1/Parkin は車のブレーキのように、普段は私達がパーキンソン病になることを防いでいますが、故障(遺伝子が変異)してそのはたらきが失われるとパーキンソン病になることがわかっています。
ではParkin/PINK1はどのようなはたらきをすることで、パーキンソン病を防いでいるのでしょうか?
(3)マイトファジー(ミトコンドリアオートファジー)とパーキンソン病
ミトコンドリアは、その細胞が生きるために必要なエネルギーを作り出したり、取り込まれた物質の代謝を行なう、非常に重要なオルガネラ(細胞内小器官)です。

しかし、人間が生きる上で晒される様々な環境的要因で傷ついたり、機能を失ってしまうことがあります。ミトコンドリアの機能が失われると、細胞にとって必要なエネルギーが作られなくなってしまうだけでなく、細胞にとって害となる物質まで排出するようになります。細胞全体の生存が脅かされる事態を避けるためには、不良なミトコンドリアを一刻も早く「取り除く」必要があります。
そこではたらくのが「マイトファジー」という、不良なミトコンドリアだけを選択して取り除くシステムです。
これまでに私たちを含めた世界中の研究グループが行ってきた研究により、
① ミトコンドリアが不良な状態になる(エネルギーを作り出すために必要な「膜電位」が失われる)と、PINK1はその表面に蓄積すること。
② PINK1はParkinを活性化させる(Parkinをはたらける状態にする)こと。
③ Parkinは、細胞内で「不要なものを分解する標識」としてはたらく “ユビキチン”という小さなタンパク質をミトコンドリア上のタンパク質に結合させるはたらきがあること。
などがわかってきました。ParkinとPINK1は互いに協力して、不良なミトコンドリア上にユビキチン鎖を作ることでマイトファジーを駆動する因子だったのです。
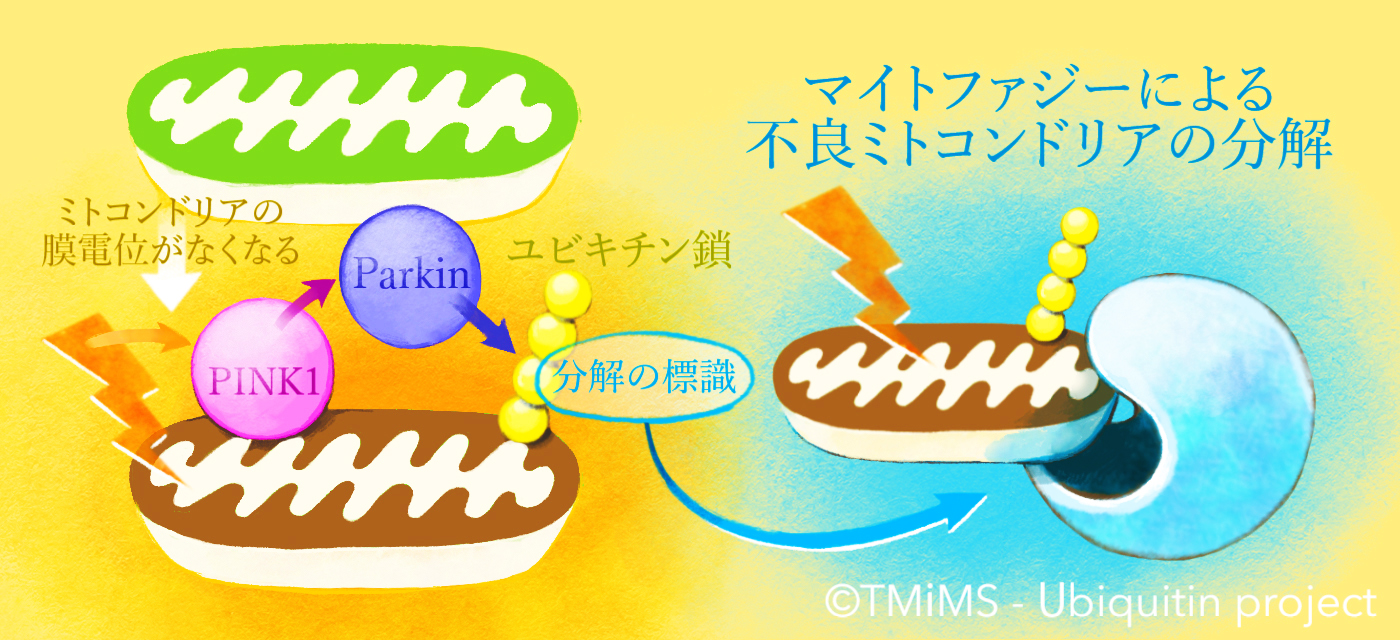
家族性のパーキンソン病ではParkin/PINK1の「故障」によってマイトファジーシステムがはたらかなくなってしまいます。その結果、神経細胞の中で不良なミトコンドリアが蓄積し、それが細胞死を引き起こして、パーキンソン病が発症すると考えられます。
(4)パーキンソン病発症メカニズムの研究と治療法開発
パーキンソン病の治療では、対症療法としてのドーパミン投与(パーキンソン病で失われる神経細胞が放出する神経伝達物質を前駆体の形で投与します)が非常に有効ではありますが、長期間使用すると様々な弊害が起こるリスクもあります。
パーキンソン病研究の長い歴史の中でもまだ明らかになっていないことはたくさんあります。しかし、ここでご紹介したようなParkin/PINK1の機能の解明は、パーキンソン病発症メカニズムを理解するための大きな一歩であり、今後さらに研究が進めばよりリスクの少ない根本的な治療が可能になるかもしれません。
現在、私たちはParkin/PINK1の機能解析に加え、Parkin/PINK1以外の家族性パーキンソン病原因遺伝子の研究や、まだ機能がわかっていない新しいマイトファジー関連タンパク質の研究、ミトコンドリア以外のオルガネラの分解システムについても積極的に取り組んでいます。将来のパーキンソン病治療に役立つことを期待して、日夜研究を続けています。
以下は理系の学生/大学院生、医者、研究者などを対象に、当研究室で行なわれている“パーキンソン病の原因遺伝子産物 PINK1 および Parkin の研究”について紹介するものです。内容はやや専門的になります。より平易な情報を必要とされる方は、<一般の方へ>のページをご覧下さい。
社会的な背景
パーキンソン病は神経伝達物質であるドーパミンを産生する黒質緻密部のドーパミンニューロンが変性・脱落することによって、線条体でのドーパミンが低下することで静止時振戦、筋強剛、動作緩慢、姿勢反射障害等の臨床症状を呈する神経変性疾患である。
(注:最近の研究からは、パーキンソン病の症状は上記のような運動障害だけではないと考えられており、病気が進行すると睡眠障害・自律神経症状・精神症状・認知機能障害などの非運動性症状が表れることも報告されている。
また変性箇所も黒質緻密部にとどまらずに、より広範にわたることが示唆されている。さらに最初に変性が起きる場所も黒質ではなく、迷走神経背側核や嗅球だとする報告もある)
本邦における患者数は 15 万人程度だといわれており、高齢者の罹患率の極めて高い難治性神経病である。家族型と孤発型に分類され、前者は遺伝性のものであり、後者は環境因子や生活環境、様々なストレスが関与していると推定されているが,いずれも発症メカニズムは完全には明らかにされていない。高齢化社会を迎えた今日、原因究明・予防および治療方法の確立が、社会的に大きく要請されている疾患である。
遺伝性パーキンソン病の原因遺伝子 PINK1 と Parkin
遺伝性パーキンソン病は孤発型パーキンソン病に比較すると患者数はずっと少ないが、原因が特定の遺伝子異常に由来することから発症機構の解明に有益であり,科学的には重要である。
その臨床症状は孤発性と完全に一致する訳ではないが、よく似ていることから、遺伝性パーキンソン病の発症原因の解明は孤発型パーキンソン病発症原因を解明するための手掛かりを与えてくれることが期待される。
現在までに10を超える遺伝子が遺伝性パーキンソン病の原因遺伝子として報告されているが、我々はその中で遺伝性劣性パーキンソン病の原因遺伝子産物 PINK1 と Parkin に着目し、「それらの機能を解析することでパーキンソン病の発症機構に迫る」という目標のもと、十数年間にわたって研究を続けてきた。PINK1 はミトコンドリア移行配列を有しており、標的蛋白質にリン酸基を導入する酵素(セリン・スレオニンキナーゼ)である。一方、Parkin は標的蛋白質にユビキチンという低分子の蛋白質を結合させる酵素(ユビキチンリガーゼ/E3)である。
ミトコンドリアとパーキンソン病
パーキンソン病の発症機構は諸説あって未だ謎に包まれているが、ミトコンドリアとの関連は古くから示唆されており、「ミトコンドリアの機能傷害がパーキンソン病の一因である」という仮説は有力である。
ミトコンドリアは細胞のエネルギー源である ATP の合成や、細胞内カルシウム濃度の制御などを行っている細胞小器官(オルガネラ)であり、ミトコンドリアとパーキンソン病の密接な関係を示唆するデータとしては(1)パーキンソン病の患者においてミトコンドリア活性の低下[特に電子伝達系の複合体I(Complex I) の機能低下]やミトコンドリアDNAの変異が観察されること、(2)MPTP やロテノンなど、ミトコンドリア機能を傷害する薬剤がヒトやモデル動物にパーキンソン病に類似した症状を引き起こすこと、などが挙げられる。
ミトコンドリアが効率良く ATP を合成するためには,内膜を隔てた膜電位(差)即ちプロトン(H+)勾配が必要である。2008年にアメリカ NIH の Youle らは、脱共役剤を用いてミトコンドリアの膜電位を失わせると Parkin が選択的に膜電位を失った損傷ミトコンドリアに移動して、マイトファジー(ミトコンドリアに対するオートファジー)経由で損傷ミトコンドリアを分解することを報告した(Narendra et al., JCB 2008)。
Parkin がミトコンドリアの品質管理に関与することはそれまでも示唆されていたが、この論文は「ミトコンドリアの膜電位」という両者の具体的な接点を示した意味で、非常に重要である。一方で、どのようにしてミトコンドリアの膜電位の異常が感知されて、何が Parkin を異常ミトコンドリアに連れて来るのかなどの点も含めて、当時は具体的なメカニズムは殆ど不明であった。
ミトコンドリア膜電位の低下による PINK1 の制御
まず PINK1 が不良ミトコンドリア処理機能のどの段階で働いているのかを明らかにするために、PINK1 欠損細胞を用いてミトコンドリアが異常になった時の Parkin の細胞内局在変化や E3 酵素活性の変化を観察した。PINK1 欠損細胞では Parkin は不良ミトコンドリアへ局在化せず、またE3酵素活性も観察できなかった。つまり、PINK1 は Parkin の上流でその局在と活性を制御する因子であることが示された(Matsuda et al., JCB 2010)。
次にミトコンドリア膜電位の低下時に、PINK1 にどのような変化が生じるのかを解析した。その結果、PINK1 がN末端のシグナル配列に依存して膜電位のある健常なミトコンドリアへ輸送されると、速やかに限定分解され、その後に細胞質のプロテアソーム依存的(= MG132感受性の)分解へと導かれることが明らかになった。
一方で、膜電位の低下はミトコンドリア内部への輸送を阻害するので、膜電位の低下時に PINK1 は分解を免れて外膜へ蓄積した。このように最初のPINK1に対する制御系として、“PINK1が量的に制御されることで、不良ミトコンドリアの出現を監視している”ことが明らかとなった(Matsuda et al., JCB 2010)。その後に蓄積した PINK1 を詳しく調べると、複合体形成や自己リン酸化といった“PINK1の質的な制御”を経て活性化されることが明らかとなった(下図)。
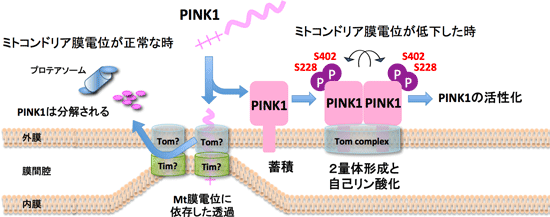
すなわち、膜電位の低下したミトコンドリアに蓄積した PINK1 は約850 kDa の巨大な複合体を形成しており、その複合体には外膜局在のミトコンドリアタンパク質輸送体 (TOM 複合体)とともに、2分子の PINK1 が含まれていることを、我々は明らかにした(Okatsu et al., JBC 2013)。さらに PINK1 は自身のキナーゼ活性に依存した自己リン酸化を受けていることが示唆された。
我々は LC-MS/MS 解析と変異体解析から、この PINK1 の自己リン酸化サイトが Ser228 と Ser402 であることを明らかにした。これらのセリンをアラニンに置換した場合、PINK1 が非リン酸化状態となるためにParkin の異常ミトコンドリアへの局在化が消失し、逆にこれらのセリンをアスパラギン酸に置換した場合、PINK1 がリン酸化模倣変異体となるために、PINK1 のリン酸化状態に依らずに Parkin を異常ミトコンドリア上に蓄積させることが可能であった(Okatsu et al., Nat. Commun. 2012)。
いくつかの PINK1 変異体では複合体形成は正常であるが、自己リン酸化に異常が生じている。このような変異体と野生型 PINK1を共発現すると変異体 PINK1 がリン酸化された。つまり、PINK1 の自己リン酸化が分子間で進行する inter-molecular 反応であるために、1つのPINK1 複合体に2分子の PINK1 が含まれている可能性が考えられた。
多数のPD患者由来の変異体 PINK1を調べたところ、上述の複合体形成や自己リン酸化のような“質的制御”に異常が観察される変異体 PINK1 が多数存在した。つまり、PINK1 疾患変異体では PINK1 が正しく活性化されないので、Parkin 依存的な不良ミトコンドリア処理機構が破綻し、その結果として遺伝性劣性PDが発症することが考えられた。
ミトコンドリア膜電位の低下と PINK1 による Parkin の制御
続けて、複合体形成と自己リン酸化によって活性化した PINK1 が、どのようにして Parkin を活性型E3に変換するのかを調べた。まず、Parkin がどのようにしてE3として機能するのかを再検討した。
2011年、RING-IBR-RING[2つの RING fingerドメインが IBR 亜鉛結合モチーフを挟みこんだドメイン構造:以下 RBR と略記]を有する RBR 型 E3 が特殊な反応中間体を経て、基質をユビキチン化することが提唱された。つまり、RBR 型 E3 は E2(ユビキチン結合酵素)からチオエステル結合型のユビキチンを受け取り、RBR 型 E3 の活性中心システインにユビキチンをチオエステル結合でリレーさせて「E3~ユビキチン反応中間体」を形成することが示唆された。
Parkin も RBR 構造を持っているが、上記の仮説が提唱された当時は Parkin もこの様式に則ったユビキチン化反応を触媒するかどうかが不明だったので、その可能性を精査した。その結果、Parkin もRING2ドメイン内にある Cys431 とユビキチンのC-末端グリシンとの間でチオエステル結合を形成し、この反応中間体を介して基質のユビキチン化反応を触媒することが明らかとなった (Iguchi et al., JBC 2013)。
次に、PINK1 が Parkin を基質としてリン酸化する可能性を検討した結果、Parkin のN末端側にある Ubiquitin-like (Ubl) ドメイン内の65番目のセリンがリン酸化を受けること、この Ser65 のリン酸化が Parkin の反応中間体(上述の Parkin Cys431とユビキチンがチオエステル結合したもの)の形成に重要であることを見出した(Iguchi et al., JBC 2013)。
しかしながら、Parkin の Ser65 リン酸化を模倣するような変異を Parkin に導入しても Parkin の活性化に対して PINK1 要求性はバイパスされないことから、Parkin 以外にも PINK1がリン酸化する重要な基質が存在することが示唆された。
その名前(Ubiquitin-like)の通りに、Parkin Ublドメインの構造はユビキチンと類似するので、次にユビキチンを解析の対象とした。Parkin と同様に、ユビキチンも65番目のセリンがリン酸化を受けることがLC-MS/MS解析と変異体解析から示された。さらに決定的に重要なことに、Parkinとユビキチンの両者に Ser65リン酸化を模倣するような変異を導入して細胞内に共発現すると、PINK1 を欠いた条件下でも Parkin の E3 酵素機能を活性化できた。
この Parkin の活性化に際して、リン酸化ユビキチンが E2 や Parkin とチオエステル結合中間体を形成する必要は無いことが示されたので、“Parkin が E3 活性を発揮する際にリン酸化ユビキチンを優先的に使用している(Parkinがリン酸化ユビキチン連結酵素として機能している)わけではない”ことが示唆された。一連の結果から、我々はリン酸化ユビキチンがリン酸化 Parkin の活性化因子として機能していると結論した (Koyano et al., Nature 2014)。
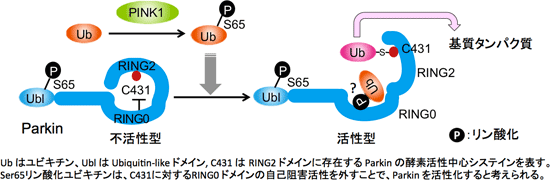
現在、我々は上図に示すようなParkin の活性化のメカニズムを考えている。2013年に海外の研究グループからParkin のX線結晶構造解析が報告されたが、その構造は「Parkin の RING0ドメインが、E3 酵素の活性中心でありユビキチンとチオエステル結合を形成する Cys431を塞ぐように位置している」ことを示していた。このことは、定常状態の Parkin が不活性型であるという我々の報告(Matsuda et al., JCB 2010)とも合致していた。
Ser65リン酸化ユビキチンがParkinの活性化因子として機能することと、上記のParkinの構造情報とを考え合わせると、Ser65リン酸化ユビキチンが Cys431 に対する RING0 ドメインの自己阻害活性を外すことによって活性中心を露出させて、Parkin が E3 機能を発揮できるように変換する可能性が高い。
多数のPD患者由来の Parkin 変異体を調べたところ、E3 活性が消失している変異体や、不良ミトコンドリアに局在できない変異体、ミトコンドリア上のタンパク質をユビキチン化できない変異体などに分類できた。つまり、PINK1と同様に、Parkin の変異体においても不良ミトコンドリア処理機構が破綻しているために PD が発症することが示唆された。
また、PINK1や Parkin に関する知見の多くは HeLa に代表される不死化培養細胞を材料に用いて得られているが、我々は初代培養神経細胞においても、ミトコンドリア膜電位低下に依存したPINK1とParkinのリン酸化、Parkin の活性型E3への変換が起きていることを確認した(Koyano et al., Genes Cells 2013)。
以上の全ての結果から、PINK1とParkin がミトコンドリア膜電位の低下というストレスに応答したミトコンドリア品質管理を担っており、その破綻が異常ミトコンドリアの増加や細胞内活性酸素種(ROS)の蓄積を引き起こして、最終的に遺伝性劣性 PD を引き起こしていることが示された(下記まとめ図を参照)。
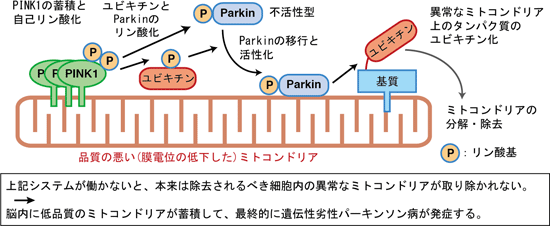
今後の課題
われわれの仕事も含めて 2010-14 年に報告された一連の論文から、若年性劣性パーキンソン病がミトコンドリアに対する品質管理の不全病であることはほぼ定説になりつつある。一方で、遺伝性優性パーキンソン病や、一般的な孤発性パーキンソン病も“膜電位を指標とするミトコンドリア品質管理の不全病”と考えてよいのかどうかは、現時点では全く不明である。
この Web site を見てわれわれの仮説に興味を持ち、その妥当性を患者由来の臨床検体などを用いて冷静に検証して下される研究者がいれば,望外の喜びである。また、われわれの仕事に興味を持たれた方も、ご連絡をいただければ幸いである。
代表的な論文
Parkin/PINK1の機能の研究ほか、松田チームの代表的な論文4報をご紹介します。
その他すべての発表論文は
<研究業績のページ>に掲載していますのでご覧ください。
(各論文の責任著者名の左に*を付けています。)
- *Matsuda N Sato S, Shiba K, Okatsu K, Saisho K, Gautier C.A, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, and *Tanaka K.
PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy.
J Cell Biol. 189: 211-221 (2010) <PubMed> - Koyano F, Okatsu K, Kosako H, Tamura Y, Go, E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon E-A, Trempe J-F, Saeki Y, *Tanaka K, and *Matsuda N.
Ubiquitin is phosphorylated by PINK1 to activate Parkin.
Nature 510: 162–166 (2014) <PubMed> - Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, *Tanaka K, and *Matsuda N.
Phosphorylated ubiquitin chain is the genuine Parkin receptor.
J. Cell Biol. 209: 111-128 (2015). <PubMed> - *Yamano K, Kikuchi R, Kojima W, Hayashida R, Koyano F, Kawawaki J, Shoda T, Demizu Y, Naito M, Tanaka K, and *Matsuda N.
Critical Role of Mitochondrial Ubiquitination and the OPTN-ATG9A Axis in Mitophagy
J. Cell Biol. 219:e201912144 (2020) <PubMed>
連絡先
松田 憲之(公益財団法人 東京都医学総合研究所:参事研究員)
matsuda-nr(a)igakuken.or.jp ((a)を@にして送信してください)

略歴
| 2001年3月 | 東京大学大学院理学系研究科生物科学修了 理学博士 |
| 2001年4月~ | 理化学研究所(和光本所)・基礎科学特別研究員 |
| 2002年4月~ | 東京都臨床医学総合研究所・外部支援研究員 |
| 2006年4月~ | 日本学術振興会・特別研究員PD |
| 2007年4月~ | 理化学研究所(横浜研究所)・上級研究員 |
| 2008年6月~ | 東京都臨床医学総合研究所(後に東京都医学総合研究所に改組)・主席研究員 |
| 現在:ユビキチンプロジェクト プロジェクトリーダー | |