Menu

公益財団法人
東京都医学総合研究所
臨床医科学研究分野
認知症研究プロジェクト
〒156-8506
東京都世田谷区上北沢2-1-6
TEL 03-6834-2349
FAX 03-6834-2349
〒156-8506
東京都世田谷区上北沢2-1-6
TEL 03-6834-2349
FAX 03-6834-2349
世界保健機関(WHO)が2012年4月に発表した推計によると、地球全体の高齢化に伴い世界全体の認知症患者が20年後に2倍、40年後には3倍に増え、2050年には100人に一人以上が認知症患者という時代になるといわれています。認知症対策に対する世界全体のコストは2010年の時点ですでに約50兆円とも言われていますが、その経済的な負担が今後ますます重くなることが容易に想像でき、少しでも発症を遅らせる治療や予防法の開発や対策が強く望まれています。
認知症と一言でいっても、脳血管の出血や梗塞による原因が明らかな血管性認知症と、原因不明の変性性の認知症に大別されます。さらに変性性認知症は、アルツハイマー型認知症(アルツハイマー病: AD)、レビー小体型認知症(dementia with Lewy bodies: DLB)、前頭側頭様変性症 (frontotemporal lobar degeneration: FTLD)、嗜銀顆粒性認知症(argyrophilic grain disease: AGD)などがあり、それぞれ臨床症状や病理が異なります。このような変性性認知症や筋萎縮性側索硬化症(ALS)をはじめとする神経難病は神経変性疾患と総称されます。神経変性疾患は、特定の細胞群が原因不明の変性をおこして脱落し、冒された脳の部位の違いによって様々な症状を呈する病気です。一部の遺伝性疾患を除いてその原因は不明ですが、多くの場合、単純に神経細胞が脱落、消滅していくのではなく、それぞれの疾患ごとに異なった異常を伴って死んでいくのが特徴です。私達はその疾患に特徴的な病変を研究することにより、発症や進行機序を究明し、病態メカニズムに基づいた治療薬や治療方法を開発していくことを目的として研究を行っています。
ADは大脳皮質の神経細胞が徐々に死んでいく病気ですが、神経原線維変化(異常タウから構成される線維性細胞内構造物)と老人斑(すなわちAβペプチドを主成分とする細胞外構造物)の二つの病理構造物を伴って変性するのが特徴です。また、DLBはレビー小体(異常αシヌクレインから構成される線維性細胞内構造物)を伴うのが特徴です。またFTLDやALSは、病型によって異なりますが、異常タウや異常TDP-43によって構成される細胞内病理構造物を伴うのが特徴です。
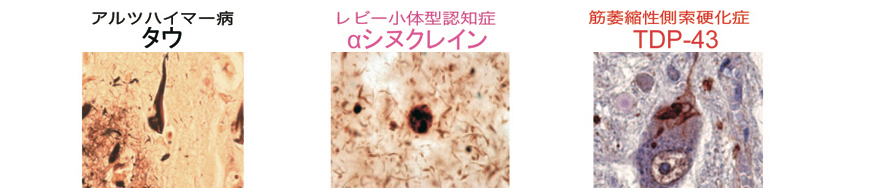
逆に死後脳の病理解析により、これらの病変が証明されないと確定診断には至りません。すなわちこれらの異常タンパク質病変は、それぞれの疾患を定義づける病理学的特徴といえます。
遺伝的にタウ、αシヌクレイン、TDP-43の遺伝子の異常をもった場合、それぞれの異常タンパク質病変を伴う変性性認知症が発症することが証明されており、これらの異常タンパク質の蓄積は単なる変性による副産物ではなく、病気の発症や進行と深い関係があります。実際、ここに挙げた細胞内異常タンパク質病変の広がりが臨床症状やその重症度と強く相関することが神経病理学的解析から明らかになっています。多数例の剖検脳のAβとタウの病変分布と臨床像の相関を調べたBraakらのADのステージ分類の仕事は有名であり、αシヌクレイン病変についてもその広がりと臨床像の関係がBraakら、Saitoらによって示されています。また最近はTDP-43の病理の進展様式についても報告がなされています。
過去30年の研究(神経病理、生化学、また分子遺伝学的解析)から、変性する部位の神経細胞やグリア細胞に疾患に特徴的な異常タンパク質を構成成分とする病変が認められること、その病変分布や広がりが患者の臨床症状と密接に関係していること、そしてそれは単なる副産物ではなく病気の発症と直接的な関連があることがわかってきました。しかしながら、なぜ特定の神経だけが冒されるのかといった「細胞特異性、選択性」や、なぜ経過とともに病状が悪化するのかという「進行性」についてはこれまで全く謎であり、ほとんど議論されてこなかったのが現状です。
私達は、ADや変性性認知症に蓄積するタウ、レビー病小体病、多系統萎縮症などに蓄積するαシヌクレイン、筋萎縮性側索硬化症(ALS)や前頭側頭葉変性症(FTLD)などに蓄積するTDP-43について、その構成成分の同定から、異常翻訳後修飾の解析を中心とした神経病理、生化学解析を行うことで、どのようなタンパク質がどのような異常をとって蓄積しているのか詳細に調べてきました。また、それぞれのタンパク質を大腸菌につくらせて、精製し、それを試験管の中で凝集するような試験管モデルを構築しました。さらには培養細胞の中で、患者脳の病態に近いタンパク質病変をつくる細胞モデルの構築を行ってきました。剖検脳の解析から、患者脳に蓄積するタンパク質は、リン酸化やユビキチン化、界面活性剤に対する不溶性、規則正しい線維構造など多くの共通点が明らかとなりました。また試験管モデルを用いた解析から、タウやαシヌクレインはリコンビナント蛋白からアミロイド様の構造、性質を有する線維が形成されること、またそれらは、異常プリオン蛋白と同様、正常分子を異常分子に変換しうる能力を有すること、異常分子の構造は多くの場合、添加したシードの構造と同じ構造に変換されることなどが示されました。さらに細胞モデルを用いた研究から、線維性異常タンパク質は条件次第で比較的簡単に細胞内に入りうること、異常タンパク質が細胞内に導入されるとそれをシードとして細胞内の同種の分子が凝集、蓄積し、患者脳に認められるリン酸化やユビキチン化などの翻訳後修飾が再現されること、凝集体が形成された細胞では細胞死や増殖抑制が観察されること、などが示されました。以下に線維化したαシヌクレインを細胞に導入したときにおこると思われる反応を模式図で示します。
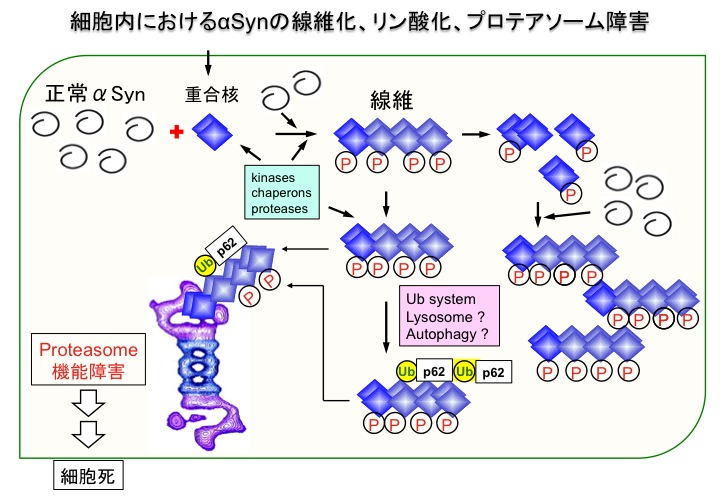
このような研究結果から、私達は、細胞内で生じた異常タンパク質が、その特殊な構造から、シナプスを介して細胞間を移動し、自身を鋳型に自己複製しながら、癌細胞やウイルスのように細胞から細胞へと伝播して広がることにより、系統的、細胞選択的な神経変性がおこり、それが徐々に進行する可能性を考えるに至り、以下のような仮説を提唱しました。
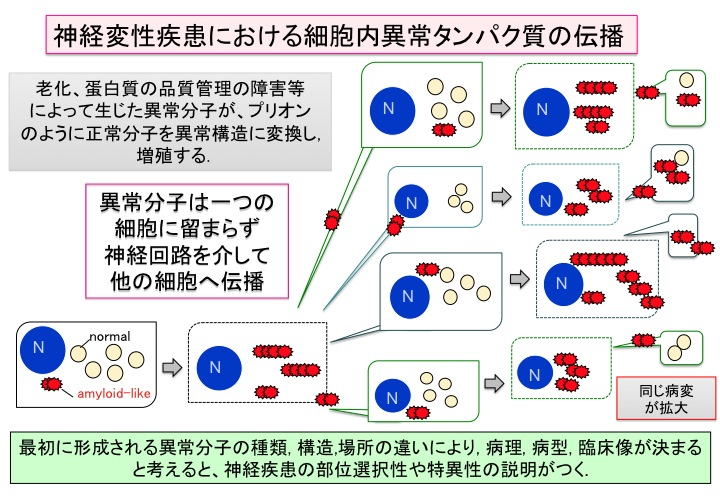
タウ、αシヌクレイン、TDP-43の異常は「細胞内」ということで、「伝播する」という考え方が常識的ではなく、議論さえされてきませんでしたが、細胞モデルの実験から、条件がととのえば異常タンパク分子が比較的簡単に細胞の中に入りうることがわかり、このように考えるようになりました。がんの場合、最初にどの臓器の細胞がどのような異常をおこしてがん化するかで、症状や進行の程度等がきまってくるのと同じように、脳のどの部位の神経細胞、グリア細胞に、どのタンパク質の、どのような構造変化が起こるかによって、病変の広がりや変性する細胞に選択性が決まり、臨床症状の違いとなって現れる可能性が考えらます。タンパク質が蓄積する細胞や変性細胞の選択性に関しては、タンパク質発現の違いが影響しますし、凝集体に対する細胞の脆弱性の違いもありますので単純ではないと思いますが、単なる拡散によって異常タンパク質が広がるのではなく、シナプスなどを介した神経回路のつながりを中心とした経路と考えらます。
神経変性疾患に認められる細胞内の異常タンパク病変については、つい最近まで「墓石を調べるようなもの」などといった批判もありましたが、家族性疾患の患者に蓄積タンパク質をコードする遺伝子の異常が多数発見されたことから、このアプローチが正しかったことが証明されました。細胞内に蓄積するタウやαシヌクレインは細胞外に蓄積するAβやプリオン蛋白と同様、「アミロイド」に特徴的なクロスβ構造とっています。アミロイドはこの構造のため、死後の分解などにも影響を受けずにそのまま残るのです。タウ、αシヌクレイン、TDP-43の異常リン酸化が死後脳で検出される理由も同じで、蓄積している分子が堅い線維構造をとっているために脱リン酸化を受けないのです。
ではどのようにして、この特殊の構造を有するタンパク質の凝集体が細胞の中で形成されるのでしょうか?これまでの研究からアミロイドの形成機構を説明するモデルとして、最初に重合核(シード)が形成され、その重合核に正常分子が結合することによって伸長反応がおこり重合が進行する「重合核依存性タンパク質重合モデル」が提唱され、実験的な証拠も累積しています。
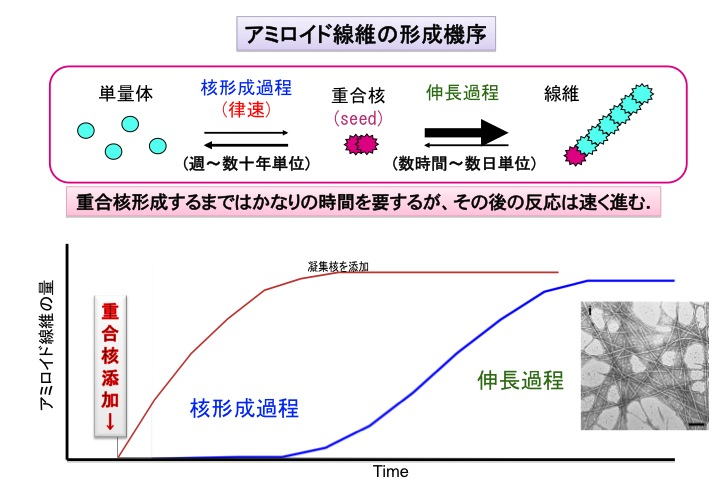
重合核が形成されるまでは長時間を有しますが、その重合核が一旦形成されると、それを核として重合反応が加速度的に進行します。また形成される線維の構造は重合核の構造や性質に依存する場合が多いことも示唆されています。
元気のよい細胞でアミロイドのような異常分子が形成されてもすぐに修正されたり、分解されたりして排除されると考えられますが、老化などによってタンパク質の品質管理システムが滞ったり、障害がおこると、そのような異常分子が形成される機会が増えると考えられます。そうなるまでには長い時間がかかると推定されますが、一度それが形成されるとその後の増殖は加速度的に進行すると考えられます。また、異常分子は細胞が死滅すると細胞外に放出され、それがエンドサイトーシスなどにより細胞の中に取り込まれ、別の細胞で増殖することが考えられます。またこのような既存のメカニズムとは全く異なる機序により異常分子が細胞間を移動する可能性も十分に考えられます。いずれにしても一旦、増殖性のアミロイド様分子が形成されるとそれが伝播して、同じ構造や性質を有する異常分子が次々と形成していく。そう考えると変性疾患の病変の広がりや進行性が説明できます。細胞は細胞膜により、その内と外を明確に仕切っており、細胞間で巨大分子が移動するはずがないと普通は考えますが、そのような「常識」にとらわれずに考えてみると納得できます。インフルエンザウイルスや狂犬病ウイルスなどはエンドサイトーシスによって細胞内に侵入することが知られていますが、異常線維構造を取ったタンパク質がその特殊な構造のゆえにウイルスと同じような性質を結果的に獲得しても不思議はなく、実験的にも実証可能です。
実際、私達は以下に示すような実験を行い、この仮説を検証しました。精製したαシヌクレインをそのまま、あるいは試験管内で振とうして線維化し、それを野生型マウスの脳内に接種するという単純な実験です。
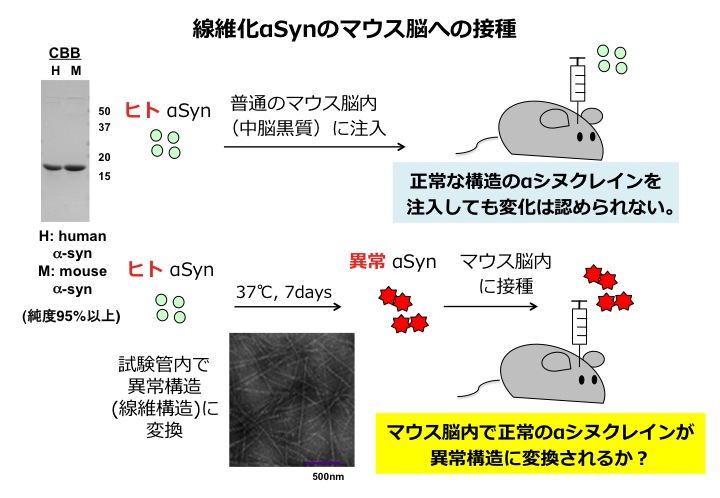
その結果、可溶性αシヌクレインを接種したマウスでは何の異常も検出されませんでしたが、線維化αシヌクレインを接種したマウスではたった3ヶ月で異常リン酸化αシヌクレイン病変が出現したのです。またその病変は時間経過と共に接種場所から遠く離れた場所にまで、ある決まったパターンをとって広がることが確認されました。 マウス脳で蓄積したαシヌクレインが接種したヒトαシヌクレインが蓄積したのか、内在性のマウスαシヌクレインが蓄積したのかを調べるため、生化学解析を行いました。その結果、接種したヒトαシヌクレインは1週間程度で消失し、3ヶ月後に蓄積してくるのは、内在性αシヌクレインであることが確認されました。この結果は、ヒト異常αシヌクレインがspecies barrierを超えマウスαシヌクレインを異常に変換することを示しています。

この接種実験により、微量の異常αシヌクレインが脳内に存在するだけで、正常なαシヌクレインが異常構造に変換され、それが急速に増幅することが証明されました。この研究で示した手法は、他の細胞内の異常タンパク質についても適用し、レビー小体型認知症をはじめとする様々な神経変性疾患の新規モデルの構築と治療法開発に応用が期待されます。このように病態基盤の理解が進めば、神経変性疾患の発症や進行の機構解明だけでなく、その予防や治療戦略が明確になると考えます。私達もこれらのモデルを使って、これまでの治療薬とは作用機序が異なる、病気の進行を止める薬剤の探索、開発を進めたいと考えています。
〒156-8506
東京都世田谷区上北沢2-1-6
TEL 03-6834-2349
FAX 03-6834-2349
Copyright © 2023 Tokyo Metropolitan Institute of Medical Science. All Rights Reserved.