

Research Center for Genome & Medical Sciences
Research Center for Genome & Medical Sciences
We study the functions of the human genome, focusing especially on transcriptional regulation. In particular, we are identifying novel genomic signals that regulate transcription in various cell types, tissues and organs. In collaboration with municipal hospitals, we will explore the molecular bases for transcriptional changes in various diseases. We will also conduct in-house collaborations with individual research groups at TMiMS to facilitate genome-scale analyses.
Research activities
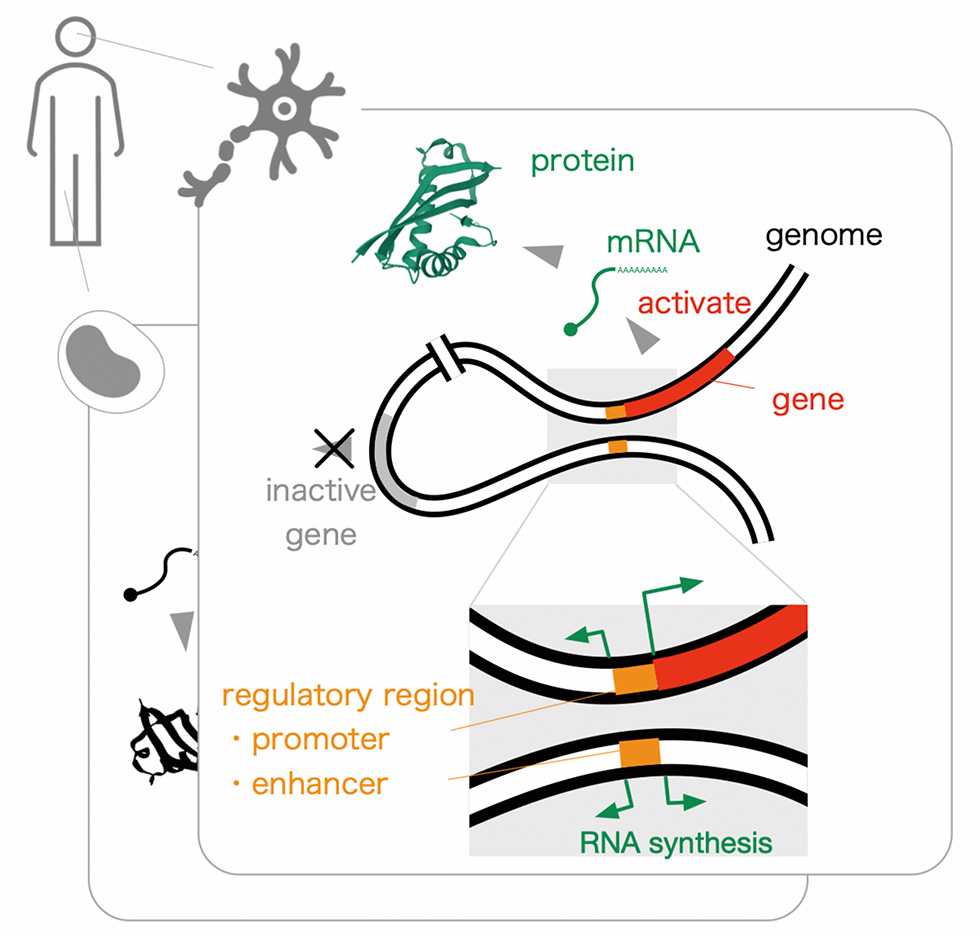
Thirty seven trillion cells in the human body carry almost identical genetic information composed of three billion bases. Within these cells, different cell types express unique sets of genes. The profiles of the transcribed genes, and the structure of their transcripts, are a unique signature of each cell type, including that derived from diseases. These profiles will serve as a molecular basis for understanding development, homeostasis and response to environmental stresses.
Transcriptional patterns are regulated by non-coding regulatory sequences that constitute nearly 99% of the human genome. We challenge to identify and dissect these regulatory segments to discover unexplored signatures and signals for cell-type and diseases-specific gene regulation. The resulting information will be used to understand diseases and to develop new diagnostics and therapeutic treatments.
Research Summary
Our body consists of around thirty-seven trillion cells, each of them carries almost identical genetic information composed of three billion base-pairs. Meanwhile, individual cells express a unique subset of genes, not all, and the expressed ones comprise the molecular basis within (or outside sometimes) the cells. Our genomes carry the structural information specifying both expressed molecules (genes), and the regulatory signals orchestrating molecules to be present in the cells (regulatory elements).
Given that such protein coding sequences occupy only 1 ~ 2% of the genome, identification of functional regions within the remaining 98 ~ 99% is crucial in understanding human biology as well as in interpretation of human diseases. Through a unique RNA profiling technology, called CAGE (Cap Analysis Of Gene Expression), that determines frequency of transcription initiation at the base-pair resolution across the genome, we discovered a series of regulatory regions, called promoters and enhancers, 10-fold or more than the protein coding genes. It indicates presence of still uncovered regulatory regions, and raises a challenge to assess their contribution to the expression of genes. We are going to tackle these challenges by combining high-throughput genome-wide experiments with large-scale computing. We will also seek the opportunities of collaborations with other research groups in TMIMS to accelerate medical science in individual fields, and with hospitals to understand diseases and to develop new diagnostics and therapeutic tools.
Selected Publications
- Jayakumar V, et al. (2021) “Chromosomal-scale de novo genome assemblies of Cynomolgus Macaque and Common Marmoset.” Sci Data. 8(1):159.
- Abugessaisa I, et al. (2021) “FANTOM enters 20th year: expansion of transcriptomic atlases and functional annotation of non-coding RNAs.” Nucleic Acids Res. 49(D1):D892-D898.
- Ito Y, et al. (2021) “Nanopore sequencing reveals TACC2 locus complexity and diversity of isoforms transcribed from an intronic promoter.” Sci Rep. 11(1):9355.
- Hirabayashi S, et al. (2019) “NET-CAGE characterizes the dynamics and topology of human transcribed cis-regulatory elements.” Nat Genet. 51(9):1369-1379.
- Yoshida, E., et al. (2017) “Promoter-level transcriptome in primary esions of endometrial cancer identified biomarkers associated with ymph node metastasis.” Sci. Rep. 7(1):14160
- Takamochi,K., et al. (2016) “Novel biomarkers that assist in accurateÝiscrimination of squamous cell carcinoma from adenocarcinoma of the ung.”BMC Cancer 16(1): 760.
- Kawaji, H., et al. (2014) “Comparison of CAGE and RNA-seq ranscriptome profiling using clonally amplified and single-molecule ext-generation sequencing.”Genome Res. 24(4):708-717.
- Forrest, A.R.R., Kawaji,H., et al. (2014) “A promoter-level mammalianÞxpression atlas.” Nature, 507(7493):462-70.