Hepatitis C virus (HCV) persists in the human body for decades and damages the liver by inducing chronic inflammations, ultimately leading to cirrhosis and liver cancer. A new study led by Daisuke Yamane, D.V.M., Ph.D. a chief researcher of Viral Infection Control Project at the Tokyo Metropolitan Institute of Medical Science and Ikuyo Ichi, an associate professor of Faculty of Core Research Natural Science Division at Ochanomizu University, has discovered how lipid metabolism in liver cell provides protection against HCV infection. Results published in Cell Chemical Biology show high levels of polyunsaturated fatty acids (PUFAs), the synthesis of which is under the control of FADS2 (fatty acid desaturase 2), is associated with the cellular resistance to HCV infection. Accumulated intracellular PUFAs undergo iron-dependent lipid peroxidation (LPO) in a manner similar to its regulation of ferroptosis, an iron-dependent form of non-apoptotic cell death, thereby down-regulating HCV replication. This study also demonstrates that pharmacological induction of iron-dependent LPO with ferroptosis inducing compounds elicits strong antiviral effects by altering the functional conformation of the viral replication machinery (a.k.a. viral replicase) to a suboptimal state.
The research team has previously demonstrated that LPO is a critical restriction factor for HCV, a leading cause of chronic liver disease (Yamane et al., Nature Medicine, 2014, 20:927-35). Endogenous LPO alters the conformation of the viral RNA replicase tethered to endoplasmic reticulum-derived membranes, impairing its ability to replicate the viral genome and affecting the susceptibility to antiviral agents. The regulatory mechanisms controlling LPO have been extensively studied particularly in the field of cancer biology since the discovery that certain types of cancer cells are more susceptible to cell death triggered by iron-dependent production of LPO, known as ferroptosis. Yet, the cellular processes that promote endogenous LPO and restrict HCV replication remained poorly characterized.
This study investigated metabolic processes that promote LPO, a key restriction factor for HCV. Although LPO can result from both enzymatic and non-enzymatic reactions, this study shows that chelation of the iron with deferoxamine abolished LPO and promoted HCV replication. This indicates that HCV replication is predominantly regulated by iron-dependent production of reactive oxygen species, resembling the mechanism triggering ferroptosis. Whereas ferroptosis has not been recognized to have an antiviral effect on any virus, noncytolytic concentrations of a ferroptosis-inducing compound, erastin, suppressed HCV replication. Mechanistically, noncytolytic concentrations of erastin disrupted HCV replicase function by altering the conformation of the membrane-anchored viral protease, enhancing its susceptibility to direct-acting antiviral drugs, and facilitating viral clearance. In contrast, ferroptosis inhibitors, such as deferoxamine and ferrostatin-1, lowered the binding affinity of these antivirals and reduced their inhibitory effects against the virus.
In an attempt to map fatty acid metabolic pathways that contribute to the production of iron-dependent LPO in hepatocytes, this study identified FADS2, which catalyzes the desaturation of fatty acids to increase the intracellular abundance of highly unsaturated PUFAs, as a master regulator that promotes LPO and suppresses HCV replication, and also as a key factor in sensitizing cells to ferroptosis. Knocking out FADS2 expression resulted in complete resistance to erastin-induced ferroptosis, whereas ectopic expression of FADS2 promoted susceptibility to ferroptosis-inducing compounds in ferroptosis-resistant cells. Importantly, publicly available data show that FADS2 expression correlates with sensitivity to erastin-induced cell death in large intestine cancer cells, indicating that FADS2 expression determines ferroptosis sensitivity in multiple cell types.
Fatty acid profiling of cultured liver cells revealed that FADS2 expression drives synthesis of Mead acid, an atypical long-chain PUFA, the synthesis of which is known to be activated by a deficiency of essential fatty acids via the compensatory pathway using oleic acid as a material, in addition to arachidonic acid generated by the canonical pathway. These results highlight the unexpected predominance of the non-canonical lipid metabolism in cultured cells widely used in laboratories.
Together, these findings demonstrate the functional importance of the FADS2-regulated PUFA biosynthesis pathway in ferroptosis, and suggest the possibility that this pathway might be pharmacologically manipulated to attenuate HCV replication.
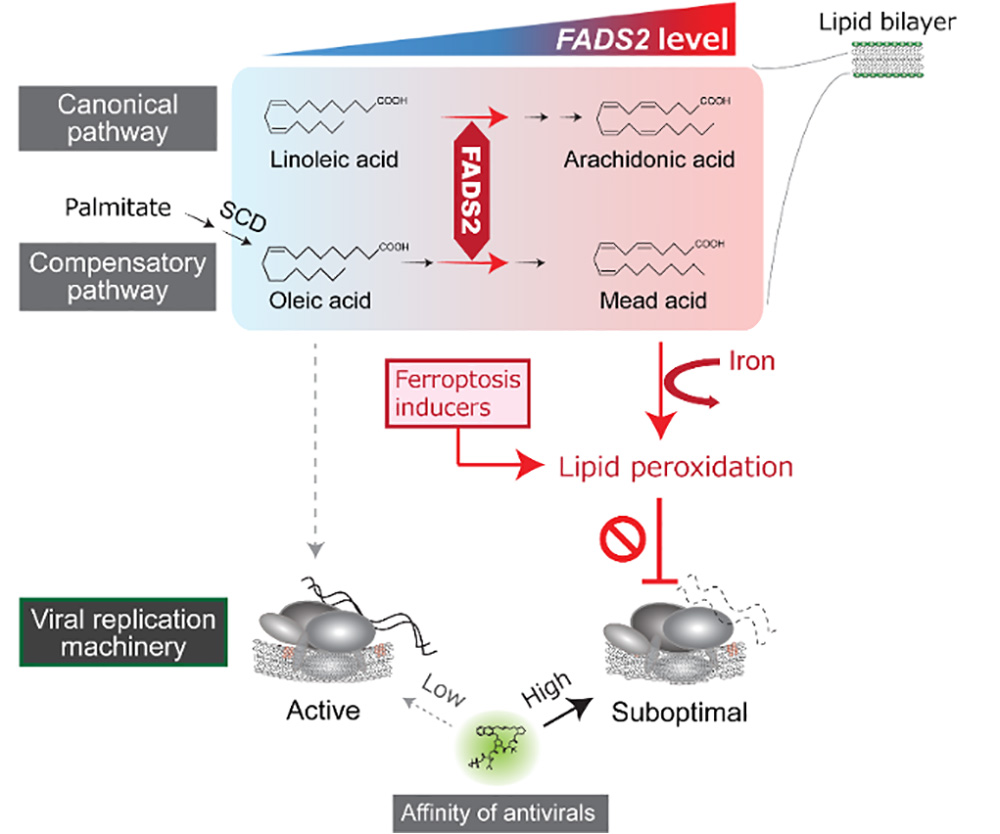
IMAGE: Model showing how FADS2-regulated fatty acid metabolism triggers lipid peroxidation and regulates hepatitis C virus replication and sensitivity to antiviral drugs.
