
TOPICS 2017
-
Naoko Niimi published a paper on “A spontaneously immortalized Schwann cell line from aldose reductase-deficient mice as a useful tool for studying polyol pathway and aldehyde metabolism” in Journal of Neurochemistry.

-
Dr. Mari Suzuki of Diabetic Neuropathy Project had won the Young Investigator Award (basic research section) of the 36th Annual Meeting of Japan Society for Dementia Research.
-
Dr. Syudo Yamasaki (Mental Health Promotion Project), received the Best Poster Award from 20th International Congress of International Society for Psychological and Social approaches to psychosis.
-
Article publication on the effectiveness of psychosocial intervention programme for challenging behaviour of people with dementia. Miharu Nakanishi (Mental Health and Nursing Research Team)
-
Yukiko Yoshida (Ubiquitin Project) published a paper on “Ubiquitination of exposed glycoproteins by SCFFBXO27 directs damaged lysosomes for autophagy” in PNAS.
-
Masato Hosokawa published a paper on “Accumulation of multiple neurodegenerative disease-related proteins in familial frontotemporal lobar degeneration associated with granulin mutation” in Scientific Reports.
-
Hikaru Tsuchiya (Laboratory of Protein Metabolism) published a paper on "Elucidation of the major pathway for proteasomal degradation" in Molecular Cell.
-
Kaori Endo (Mental Health Promotion) published a paper on "preference
for solitude, social isolation, and suicidal problems in adolescence" in Journal of Adolescent Health.
-
Phosphatidylethanolamine dynamics are required for osteoclast fusion Scientific Reports, Atsushi Irie.
 Scientific Reports 7, Article number: 46715 (2017)
Scientific Reports 7, Article number: 46715 (2017)
-
Hisao Masai (Vice Director-general) received the 2017 Prize for MEXT Science and Technology (Research Category).
-
Minoru Saitoe (Learning and Memory Project), is selected for winner of 19th Tokizane Prize.
HOME > Topics2017 > 4 May 2017
4 May 2017
Masato Hosokawa(Dementia Research Project) published a paper on “Accumulation of multiple neurodegenerative disease-related proteins in familial frontotemporal lobar degeneration associated with granulin mutation” in Scientific Reports.
Granulin mutations causing progranulin reduction may accelerate the intracellular accumulation of neurodegenerative-related proteins other than TDP-43 in the brains of familial frontotemporal lobar degeneration patients with granulin mutations - the pathologies seen in GRN mutation cases may possibly be renamed “neuronoglial multiple proteinopathies”
- <Journal>
- Scientific Reports, 7(1): 1513, 2017 May 4
doi: 10.1038/s41598-017-01587-6. - <Authors>
- Masato Hosokawa、Hiromi Kondo、Geidy E Serrano、Thomas G Beach、Andrew C Robinson、David M Mann、Haruhiko Akiyama、Masato Hasegawa、Tetsuaki Arai
- <Title>
- Accumulation of multiple neurodegenerative disease-related proteins in familial frontotemporal lobar degeneration associated with granulin mutation
https://www.nature.com/articles/s41598-017-01587-6
1. Background
Mutations in the granulin gene were identified in patients with familial Frontotemporal Lobar Degeneration in 2006. Granulin transcript haploinsufficiency has been proposed as a disease mechanism that leads to the loss of functional progranulin protein. Granulin mutations were initially found in tau-negative patients, though recent findings indicate that these mutations are associated with other neurodegenerative disorders with tau pathology, including Alzheimer’s disease and corticobasal degeneration. Moreover, a reduction in progranulin in tau transgenic mice is associated with increasing tau accumulation (Hosokawa M et al, Journal of Neuropathology and Experimental Neurology 74: 158-165, 2015). To investigate the influence of a decline in progranulin protein on other forms of neurodegenerative-related protein accumulation, human granulin mutation cases were investigated by histochemical and biochemical analyses.
2. Research summary
Results showed a neuronal and glial tau accumulation in granulin mutation cases. Tau staining revealed neuronal pretangle forms and glial tau in both astrocytes and oligodendrocytes. Furthermore, phosphorylated α-synuclein-positive structures were also found in oligodendrocytes and the neuropil. Immunoblot analysis of fresh frozen brain tissues revealed that tau was present in the sarkosyl-insoluble fraction, and composed of three- and four-repeat tau isoforms, resembling Alzheimer’s disease.
Figure 1. Tau accumulation in GRN mutation cases
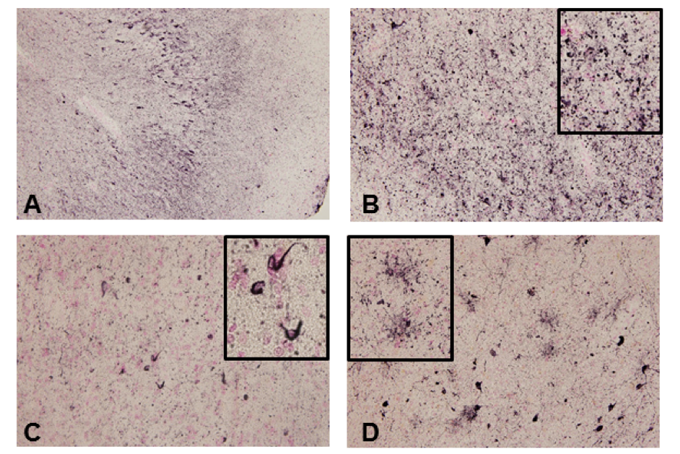
(A) Phosphorylated tau accumulation in neurons. (B) In the neuropil, fine tau–positive granules were abundant. (C) In the white matter, tau-positive oligodendroglial coiled bodies were observed. (D) Phosphorylated tau accumulation in astrocytes.
Figure 2. α-synuclein accumulation in GRN mutation cases
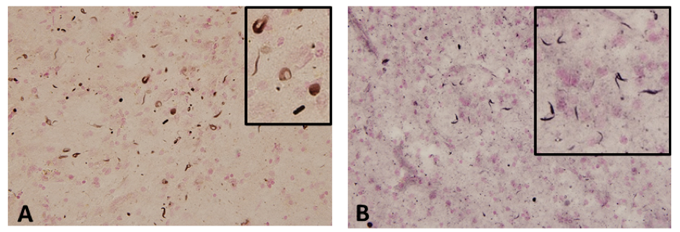
(A, B) Phosphorylated α-synuclein accumulation in the temporal lobe of GRN mutation cases.
3. Conclusion
The results of the present study show that GRN mutations causing PGRN reduction may accelerate the intracellular accumulation of not only TDP-43 but also tau and α-synuclein in the brains of familial FTD patients with GRN mutations. This suggests that GRN mutations causing PGRN reduction may be causative or represent risk factors for multiple proteinopathies (TDP-43, tauopathy or α-synucleinopathy). Our findings suggest that the pathologies seen in GRN mutation cases may possibly be renamed “neuronoglial multiple proteinopathies”.
This research was partially supported by a Daiwa Securities Research Foundation grant to M. Hosokawa.
- <Contact>
- Dementia Research Project, Department of Dementia and Higher Brain Function,
Tokyo Metropolitan Institute of Medical Science - Chief Researcher : Masato Hosokawa
- Email : hosokawa-ms [at] igakuken.or.jp