Replication checkpoint is essential for maintaining the genome integrity in response to exposure to various replication stress as well as during the normal growth. The evolutionally conserved ATR-Claspin-Chk1 pathway is induced during the replication checkpoint activation. Cdc7 kinase, required for initiation of DNA replication at replication origins, has been implicated in checkpoint activation but how it is involved in this pathway has not been known. Here, we show interaction of Claspin with Chk1 is abrogated in cancer cells, when Cdc7 expression is reduced (Cdc7low). Checkpoint activation is compromised in the APDE/A mutant of Claspin, that cannot recruit Cdc7 and thus cannot be phosphorylated by Cdc7, suggesting that Claspin may be a target of Cdc7 for checkpoint activation. Indeed, we show that Cdc7 phosphorylates CKBD (Chk1 binding domain) of Claspin, promoting the interaction between Claspin and Chk1. The residual Chk1 activation in Cdc7low cells is lost upon further depletion of casein kinase 1 (CK1γ1), previously reported to phosphorylate CKBD. Thus, Cdc7, in conjunction with CK1γ1, facilitates the interaction between Claspin and Chk1 through phosphorylating CKBD. We also show that, whereas Cdc7 is predominantly responsible for CKBD phosphorylation in cancer cells, CK1γ1plays a major role in non-cancer cells, providing rationale for targeting Cdc7 for cancer cell-specific cell killing.
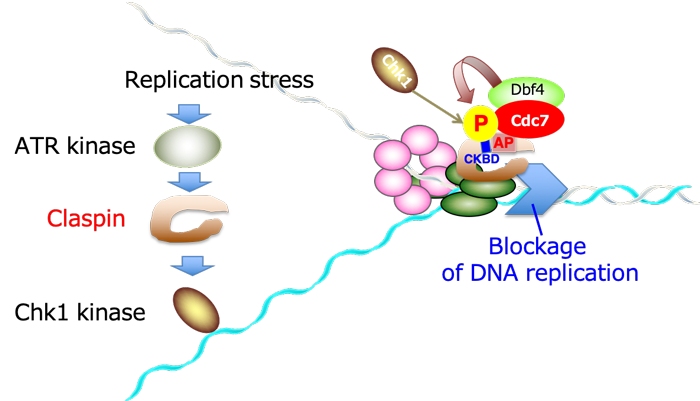
Upon replication stress, Cdc7 kinase is recruited to AP (acidic patch) on the Claspin molecule and phosphorylates CKBD present adjacent to AP. The phosphorylated CKBD now recruits Chk1 kinase and is activated to elicit downstream effector events that protect the genome from its destabilization and potentially hazardous damages.
