Dr. Makoto Saito, Dr. Fumihiko Yasui, Dr. Tsubasa Munakata, Dr. Daisuke Yamane, Dr. Kenzaburo Yamaji, Dr. Naoki Yamamoto, Dr. Ai Ikejiri, Dr. Tomoko Honda, Dr. Takahiro Sanada, and Dr. Michinori Kohara in the Viral Infection Control Project discovered that macrocyclic peptides exhibit antiviral effects against influenza virus HA and prevent pneumonia in animal models. These studies were published in Nature Communications.
The influenza A virus is a great public health issue. Most anti-influenza drugs currently used, such as oseltamivir and zanamivir, inhibit the enzymatic activity of neuraminidase (NA). NA inhibitor-resistant viruses, however, have already been isolated among seasonal H1N1, pandemic (H1N1) 2009, and even highly pathogenic avian H5N1 viruses. These resistant viruses can only be controlled by a new antiviral with a mechanism of action that is totally different from NA inhibition. The recent development of the Random non-standard Peptides Integrated Discovery (RaPID) system enables the identification of high-affinity ligands for a protein of interest from easily prepared libraries consisting of over a trillion macrocyclic peptides. Here we report bi-functional, wide tropic macrocycles that bind the influenza viral envelope protein hemagglutinin and inhibit virus infection by blocking adsorption and fusion. They showed powerful efficacy in preventing severe pneumonia at later stages of infection in mouse and non-primate cynomolgus macaque models.
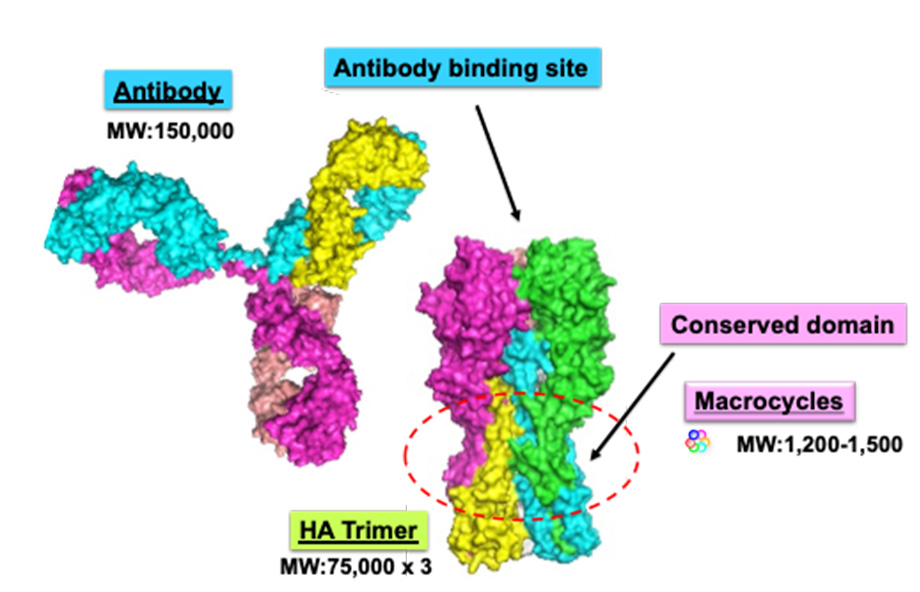
Hemagglutinin (HA) is one of the envelope proteins of influenza virus, and HA-targeted antibodies can neutralize HA-mediated virion-cell binding. To devise smaller molecules capable of binding to the influenza viral HA as potential antiviral agents, we used an emerging technology called the Random non-standard Peptides Integrated Discovery (RaPID) system. This technology allowed us to express thioether-macrocyclic peptides in a custom-made in vitro translation system and display this massive library (greater than 1012 members) on cognate mRNA templates. Iterative selection rounds were then performed to enrich potent macrocyclic binders against recombinant HA derived from the highly pathogenic avian influenza virus. Of the 28 candidates, eight iHA macrocycles (iHA-11, -12, -14, -18, -19, -23, -24, and -100) inhibited plaque formation of the low pathogenic H5N1 avian virus A/duck/Hokkaido/Vac-3/07 (H5N1/Vac-3). Among these macrocycles, iHA-24 and iHA-100 remarkably reduced the plaque number of H5N1/Vac3 and size, and also inhibited plaque formation against different antigenicities among clades of H5N1 viruses: A/Vietnam/UT3040/04 (H5N1/Vietnam; clade 1), A/whooper swan/Mongolia/3/05 (H5N1/Mongolia; clade 2.2), and A/whooper swan/Hokkaido/1/08 (H5N1/Hokkaido; clade 2.3.2.1).
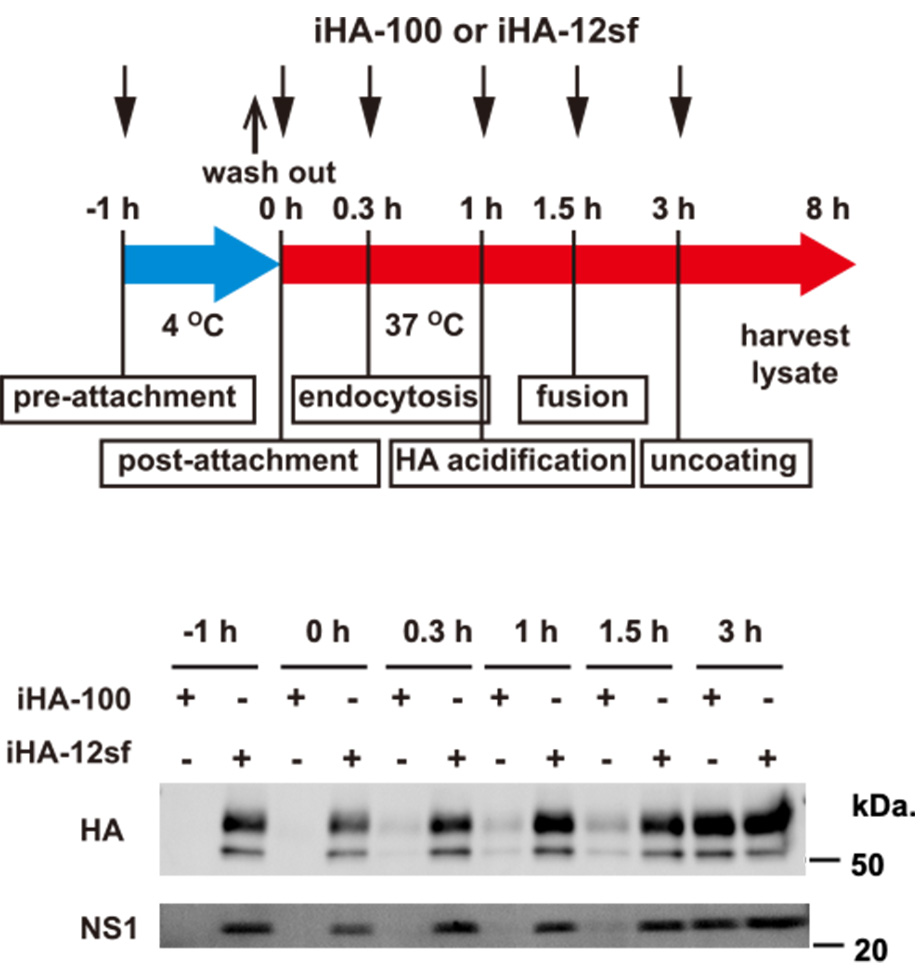
To further examine the target of iHA-100 after adsorption, iHA-100 was treated at the virus infection steps of uncoating and fusion. Virus replication was inhibited by iHA-100 treatment at −1 h, 0 h, 0.3 h, 1 h, and 1.5 h after infection, but uncoating at 3 h after infection was not inhibited. The negative control peptide iHA-12sf, which has a scrambled iHA-12 amino acid sequence, did not inhibit virus entry. Thus, the fusion step may be a critical target of iHA-100.
We evaluated the in vivo efficacy of iHA-100 against influenza virus infection in a murine lethal infection model. H5N1-infected mice were intranasally administered 1.9 mg/kg/day iHA-100 at 0 (3 h), 2, 4, or 6 days after infection. The survival rate in the iHA-100-treated group was comparable with the survival rate in the zanamivir-treated group. Although all mice in the zanamivir-treated group died due to severe weight loss when administration was delayed to 4-8 days post-infection (dpi) and 6-10 dpi, the iHA-100-treated group exhibited slight weight loss, eventually recovered, and showed 40% survival.
We further evaluated the in vivo efficacy of iHA-100 against influenza virus infection in a non-human primate cynomolgus macaque infection model. The average body temperature increase in the iHA-100-treated group was lower than that in the vehicle group. Regarding pathological findings, lung weights were relatively lower in the iHA-100-treated group than in the vehicle-treated group. In nasal, oral, and bronchial swabs of vehicle-treated monkeys, virus was detected until 6 dpi. In contrast, virus in these swabs of iHA-100-treated monkeys disappeared at 3-6 dpi.
In this work, to devise smaller molecules capable of binding to influenza viral HA as potential antiviral agents, we use the RaPID system to obtain HA-targeting macrocycles, named iHAs. Among candidate iHAs, iHA-24 and iHA-100 show inhibitory effects on the in vitro replication of a wide range of Group 1 influenza viruses. Notably, iHA-100 bifunctionally inhibited viral adsorption and membrane fusion, both of which are mediated by HA. The binding site for iHA-100 in HA is the stalk domain, which is highly conserved among various subtypes of influenza virus. Moreover, iHA-100 exhibits efficacy in inhibiting virus growth and preventing severe pneumonia at later stages of infection in mouse and non-human primate cynomolgus macaque models.