Researchers from the Calpain group— Dr. Chihiro Hisatsune and Project Leader (then) Dr. Yasuko Ono— have clarified how mutations outside the catalytically essential protease core region of calpain-3 cause LGMDR1. The research group identified two mutually independent pathways: (1) Reduction of enzymatic activity due to impaired oligomerization, and (2) Mislocalization within skeletal muscle cells due to weakened interaction with titin, a giant cytoskeletal protein. These findings are expected to deepen our understandings of the pathogenesis of LGMDR1 and contribute to the development of fundamental therapies for LGMDR1 in the future.
This study was published in the online edition of the American scientific journal Journal of Biological Chemistry on February 11, 2026.
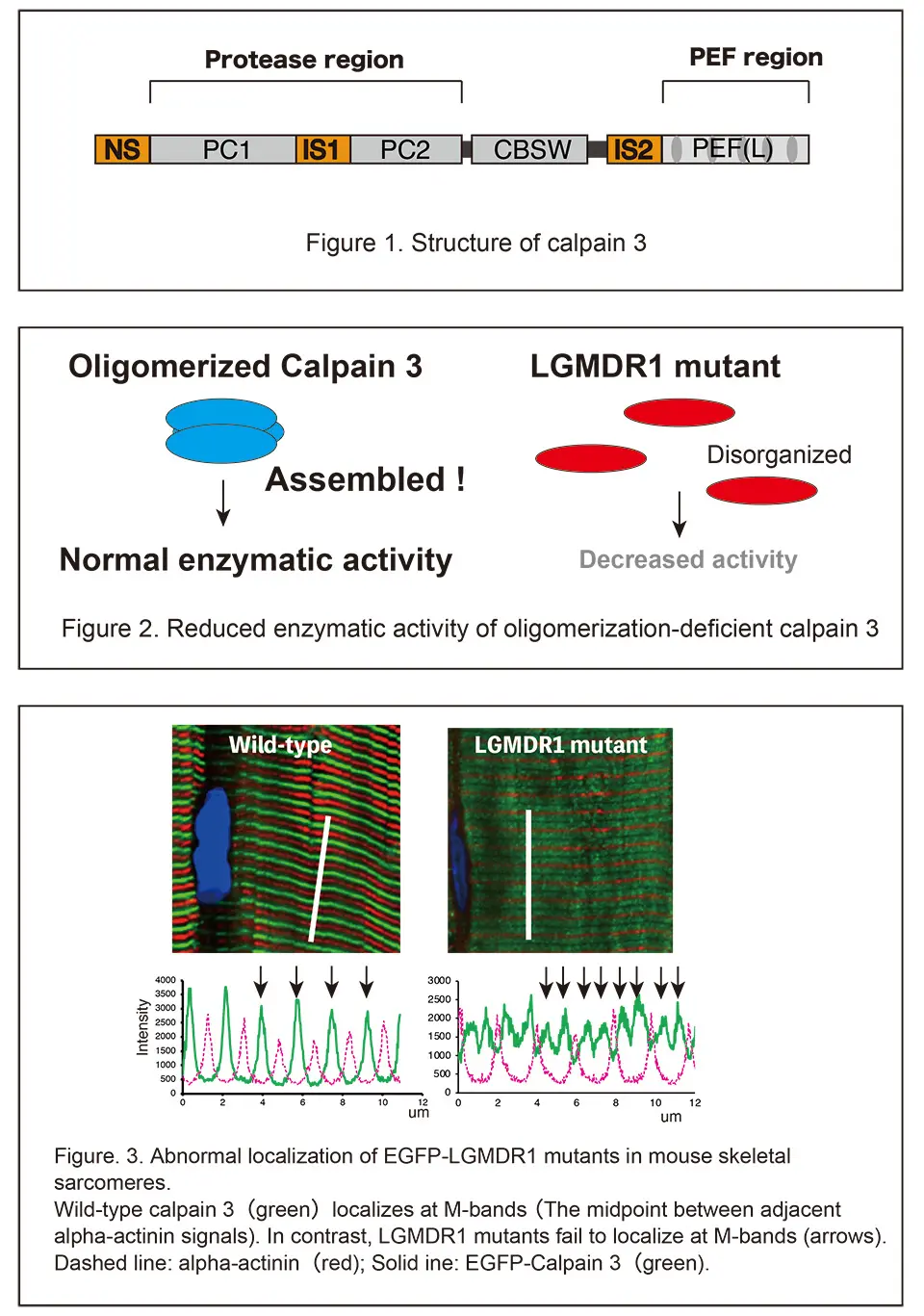
Limb-Girdle Muscular Dystrophy R1 (LGMDR1) is a rare disease characterized by the progressive weakening of muscles close to the trunk of body (proximal limb muscles). Since CAPN3 which encodes calpain-3—a calcium-dependent cysteine protease—was identified as the causal gene for LGMDR1, its biochemical characteristics have been studied extensively. However, with mutations being distributed all over the gene even outside the exons corresponding to catalytic region, it remained unknown why mutations in such diverse regions all lead to the pathology.
In the present study, the group focused on genetic mutations found in the penta-EF hand (PEF) domain located at the COOH-terminus of calpain-3.
Their results revealed two independent pathogenic routes:
The team found that some mutations in the PEF domain inhibit the binding of calpain-3 molecules to one another (oligomer formation). This failure of oligomerization causes a decrease in enzymatic activity, i.e., its function as a protease.
By expressing LGMDR1 mutants in the tibialis anterior muscle of mice using Adeno-Associated Virus (AAV), the researchers observed that certain mutants could not reside at calpain-3’s correct intracellular location—the M-line of the muscle sarcomere—due to weakened association with titin, a giant cytoskeletal protein. Importantly, this mislocalization occurred irrespective of their oligomerization status. Furthermore, the group successfully ameliorated to correct localization of a certain mutant by forcing it to form oligomers with wild-type calpain-3.
This study demonstrates that LGMDR1 mutations within the PEF domain of calpain-3 trigger the pathology through two distinct pathways: decreased enzymatic activity via impaired oligomerization, and abnormal subcellular localization independent of oligomerization. Investigating how mutations in other regions affect oligomerization will provide clearer insights into the pathogenesis of LGMDR1. In addition, investigating the structure of muscle sarcomeres expressing LGMDR1 mutants at the electron microscope level would reveal the mechanism by which calpain-3 underpins the structural integrity of the sarcomere. The present discovery that wild-type proteins can rescue mutant localization supports the validity of AAV-mediated calpain-3 gene therapy, marks a significant step toward developing effective treatments in the future.
This research was supported by Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (C) (Chihiro Hisatsune, Grant Number 22K07014 and 25K10312), Grants-in-Aid for Scientific Research (C) (Yasuko Ono, Grant Number 22K06156).