Researches
Two Loci Contribute to Age-Related Hearing Loss Resistance in the Japanese Wild-Derived Inbred MSM/Ms Mice
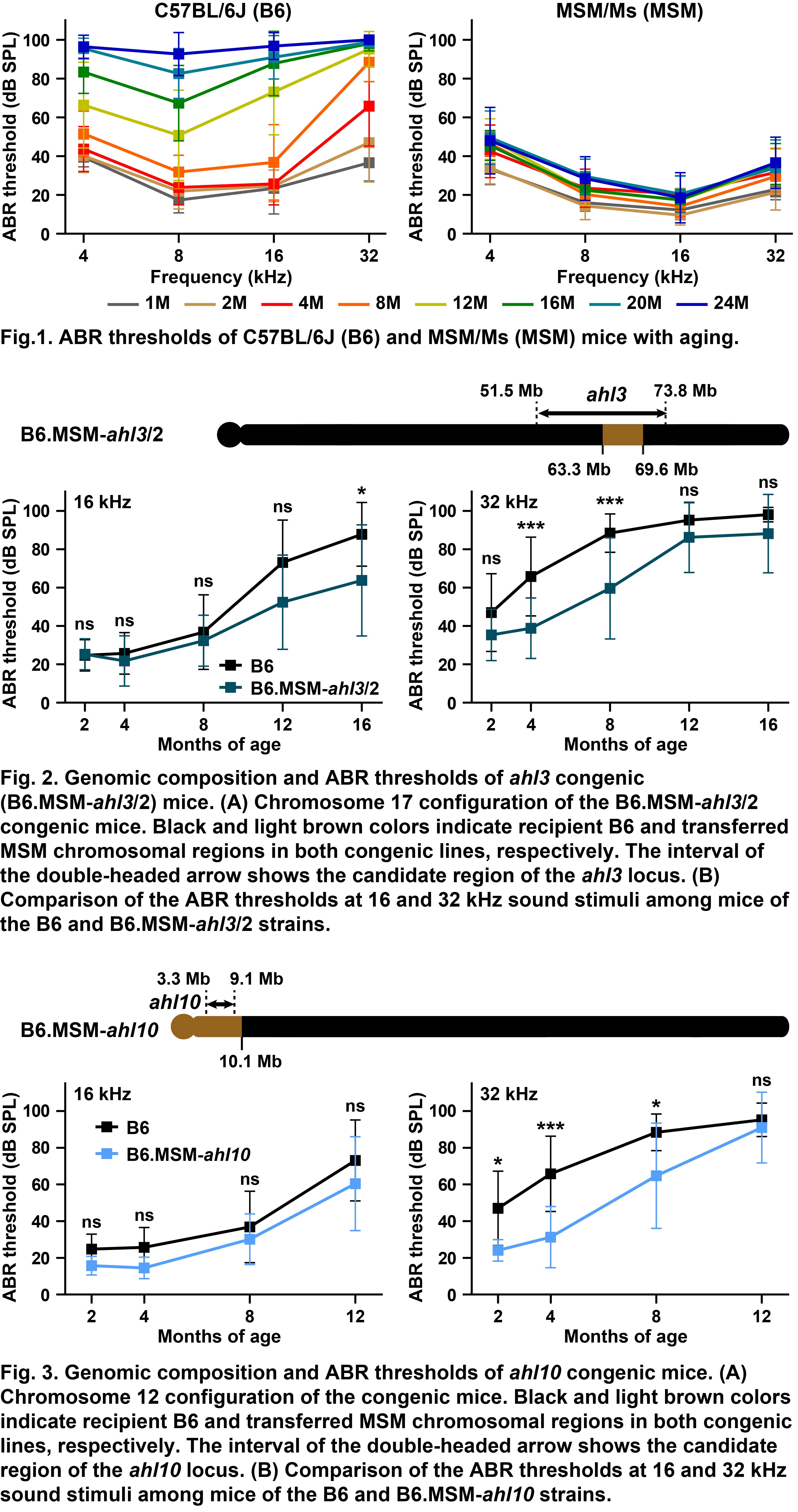
An MSM/Ms strain was established using Japanese wild mice, which exhibit resistance to several phenotypes associated with aging, such as obesity, inflammation, and tumorigenesis, compared to common inbred mouse strains. MSM/Ms strain is resistant to age-related hearing loss, and their auditory abilities are sustained for long durations (Fig. 1). The age-related hearing loss 3 (ahl3) locus contributes to age-related hearing in MSM/Ms strain. We generated ahl3 congenic strains by transferring a genomic region on chromosome 17 from MSM/Ms mice into C57BL/6J mice. Although C57BL/6J mice develop age-related hearing loss because of the ahl allele of the cadherin 23 gene, the development of middle- to high-frequency hearing loss was significantly delayed in an ahl3 congenic strain (Fig. 2). Moreover, the novel age-related hearing loss 10 (ahl10) locus associated with age-related hearing resistance in MSM/Ms strain was mapped to chromosome 12. Although the resistance effects in ahl10 congenic strain were slightly weaker than those in ahl3 congenic strain, slow progression of age-related hearing loss was confirmed in ahl10 congenic strain despite harboring the ahl allele of cadherin 23 (Fig. 3). These results suggest that causative genes and polymorphisms of the ahl3 and ahl10 loci are important targets for the prevention and treatment of age-related hearing loss.
This work was supported by JSPS KAKENHI grant numbers 15H04291, 21K19196, 21K09643, and 14J06119. This research was also supported by AMED-CREST under grant number 22gm1510004.
Yasuda et al. 2022 Biomedicines 10, 2221.
Myosin VI Haploinsufficiency Reduced Hearing Ability in Mice
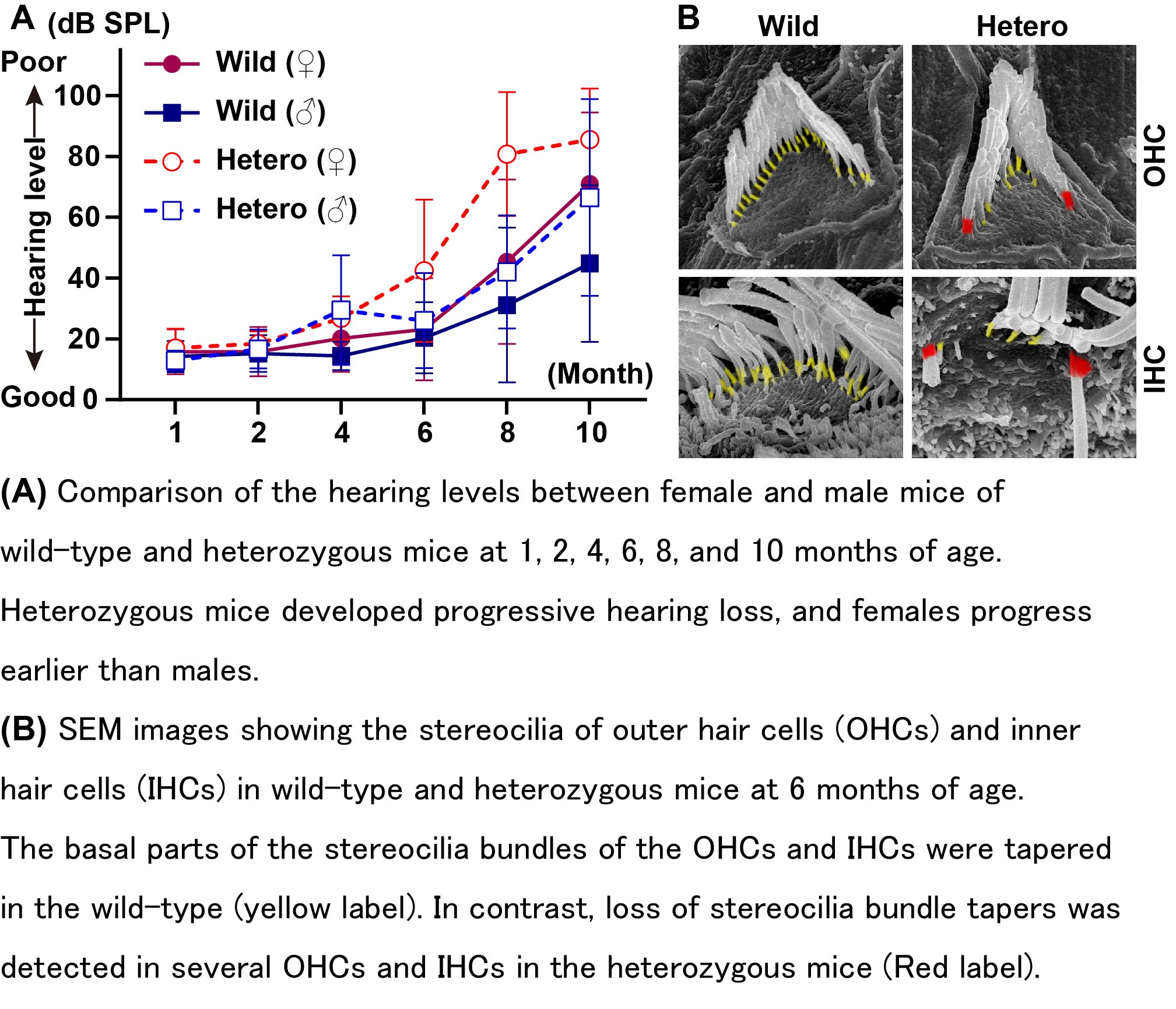
In human, myosin VI (MYO6) haploinsufficiency causes postlingual progressive hearing loss. Because the usefulness of mouse models remains unclear, we produced novel Myo6 null (-/-) mutant mice and analyzed the hearing phenotypes of Myo6+/- (+/-) heterozygous mutants. We first recorded and compared the auditory brainstem responses and distortion product otoacoustic emissions in control Myo6+/+ (+/+) wild-type and +/- mice. These hearing phenotypes of +/- mice were mild; however, we confirmed that +/- mice developed progressive hearing loss. In particular, the hearing loss of female +/- mice progressed faster than that of male +/- mice. The stereocilia bundles of +/- mice exhibited progressive taper loss in cochlear inner hair cells (IHCs) and outer hair cells (OHCs). The loss of OHCs in +/- heterozygotes occurred at an earlier age than in +/+ mice. In particular, the OHCs at the basal area of the cochlea were decreased in +/- mice. IHC ribbon synapses from the area at the base of the cochlea were significantly reduced in +/- mice. Thus, our study indicated that MYO6 haploinsufficiency affected the detection of sounds in mice, and we suggest that +/- mice with Myo6 null alleles are useful animal models for gene therapy and drug treatment in patients with progressive hearing loss due to MYO6 haploinsufficiency.
This work was supported by JSPS KAKENHI for Challenging Exploratory Research (JP15K15625) and Grant-in-Aid for Young Scientists (JP18K16913) and (B) (JP15K18393).
Seki et al. 2021 Neuroscience 478, 100-111.
c.753A>G genome editing of a Cdh23ahl allele delays age-related hearing loss and degeneration of cochlear hair cells in C57BL/6J mice
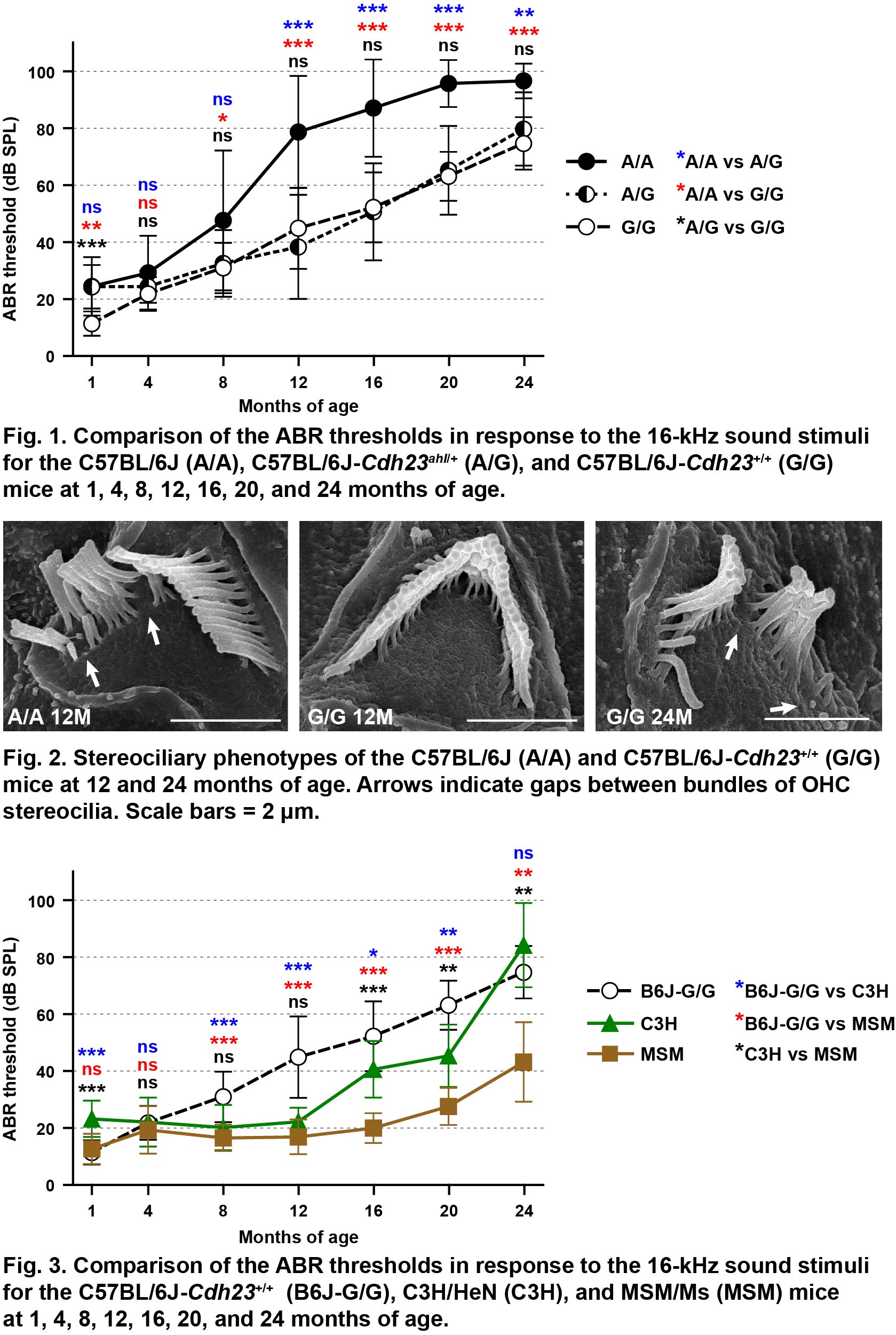
We investigated the hearing phenotypes of the C57BL/6J and genome-edited C57BL/6J-Cdh23+/+ (c.753G/G) mice until 24 months of age. Comparing to the hearing levels of C57BL/6J, it of the C57BL/6J-Cdh23+/+ mice maintained good hearing levels until 14 months of age. However, the hearing levels of the C57BL/6J-Cdh23+/+ mice gradually declined and severe age-related hearing loss developed with increasing age because the stereocilia bundles exhibited abnormal phenotypes. Details in here
This work was supported by JSPS KAKENHI for Scientific Research (B) (JP23300160 and JP15H04291) and (C) (JP18K09364).
Yasuda et al. 2020 Hear Res 389, 107926.
OHC-TRECK: A Novel System Using a Mouse Model for Investigation of the Molecular Mechanisms Associated with Outer Hair Cell Death in the Inner Ear
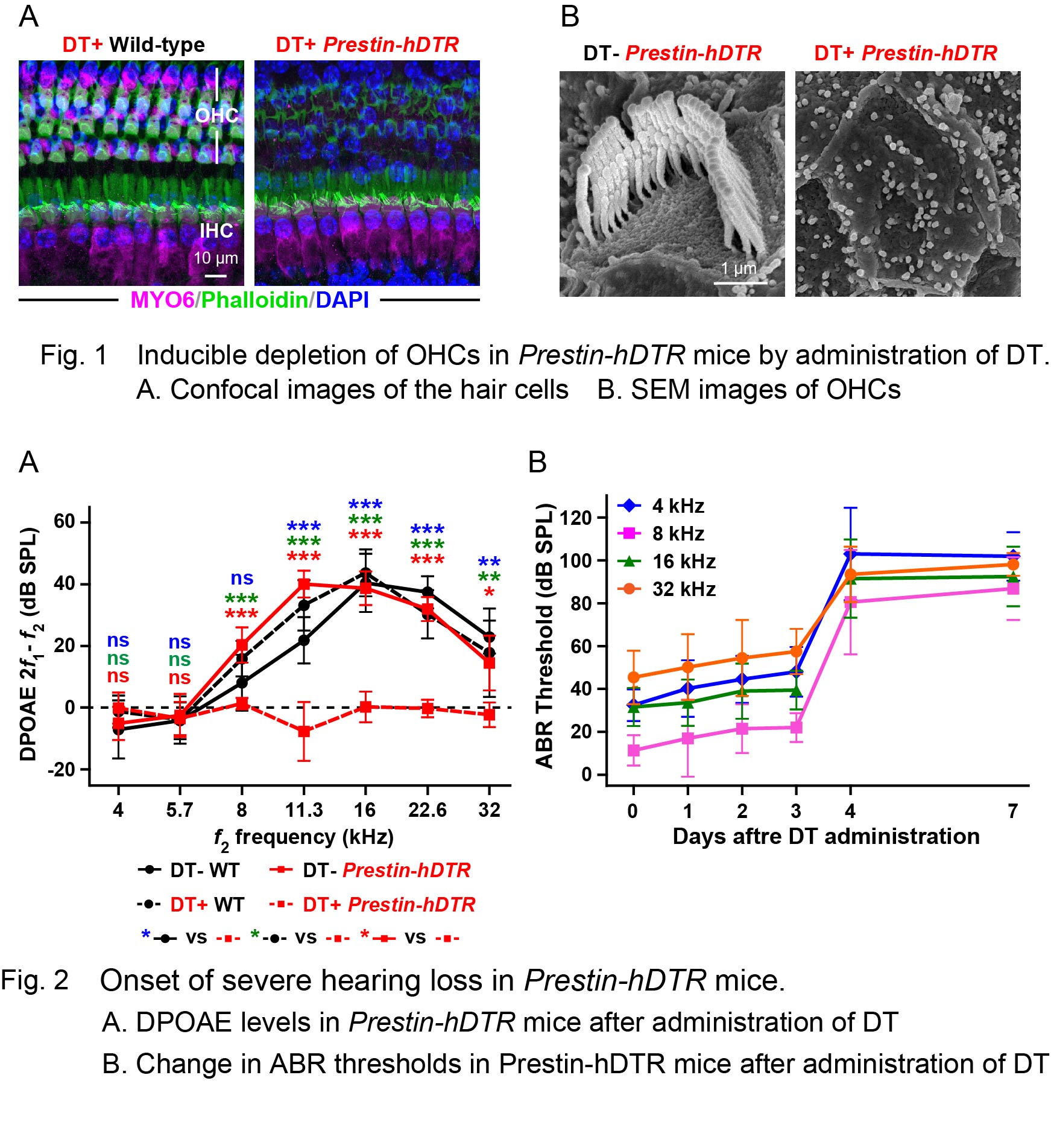
Outer hair cells (OHCs) are responsible for the amplification of sound, and the death of these cells leads to hearing loss. Although the mechanisms for sound amplification and OHC death have been well investigated, the effects on the cochlea after OHC death are poorly understood. To study the consequences of OHC death, we established an OHC knockout system using a novel mouse model, Prestin-hDTR, which uses the prestin promoter to express the human diphtheria toxin (DT) receptor gene (hDTR). Administration of DT to adult Prestin-hDTR mice results in the depletion of almost all OHCs without significant damage to other cochlear and vestibular cells, suggesting that this system is an effective tool for the analysis of how other cells in the cochlea and vestibula are affected after OHC death. To evaluate the changes in the cochlea after OHC death, we performed differential gene expression analysis between the untreated and DT-treated groups of wild-type and Prestin-hDTR mice. This analysis revealed that genes associated with inflammatory/immune responses were significantly upregulated. Moreover, we found that several genes linked to hearing loss were strongly downregulated by OHC death. Together, these results suggest that this OHC knockout system is a useful tool to identify biomarkers associated with OHC death.
This work was supported by JSPS KAKENHI for Scientific Research (C) (JP24500502 and JP15K06818) and (B) (JP16H04688) and the KAC 35th anniversary grant.
Matsuoka et al. 2019 Sci Rep 9, 5285.
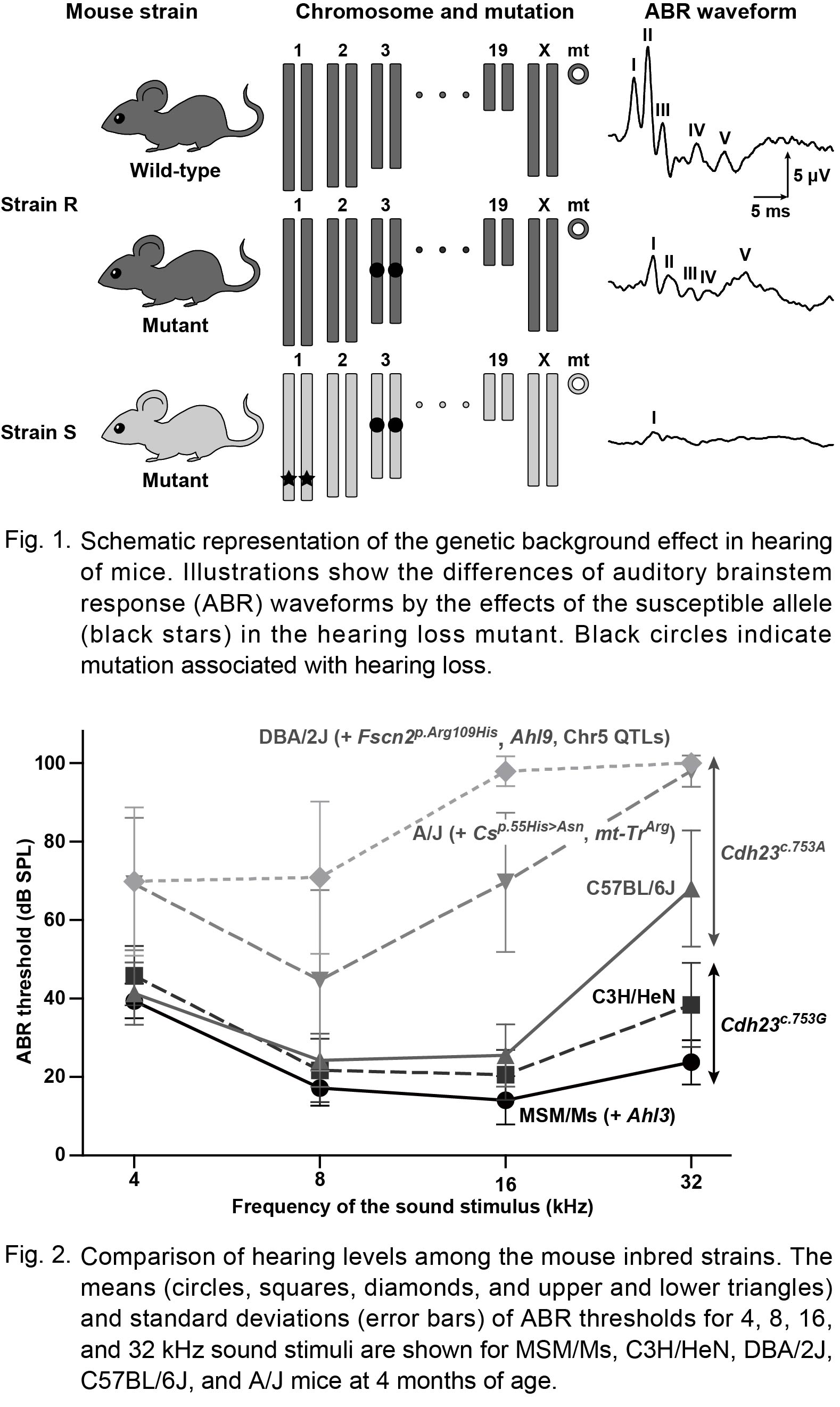
Effects of Genetic Background on Susceptibility and the Acceleration of Hearing Loss in Mice
Acquired hearing loss, which includes age-related hearing loss and noise-induced hearing loss, is a common hearing impairment and shows phenotypic variability. One reason for phenotypic variability is influence of genetic background. The modifiers underlying genetic background are modulated and advance the hearing phenotypes through gene-gene interactions with other etiological genetic factors. Moreover, the modifiers play a role in the susceptibility of environmental hearing risk factors, namely, the strength and weakness of environmental susceptibility often modulate and advance hearing phenotypes via gene-environment interactions. The complicated gene-gene and gene-environment interactions make genetic analysis of acquired hearing loss difficult. In particular, the effects of environmental factors cannot be completely excluded or controlled. Although genome-wide approaches to identify genetic modifiers have proven challenging in humans, the responsible genes and mutations are widely unknown. In this chapter, we suggest that mouse models are useful for studying genetic background effects for acquired hearing loss. The genetic analysis of mouse models identified the genetic modifiers. We review the genetic research in mouse models for acquired hearing loss to identify and confirm the modifiers by both forward and reverse genetics approaches.
Yasuda et al. 2018 In An Excursus into Hearing Loss, IntechOpen, London., UK, pp. 3–23.
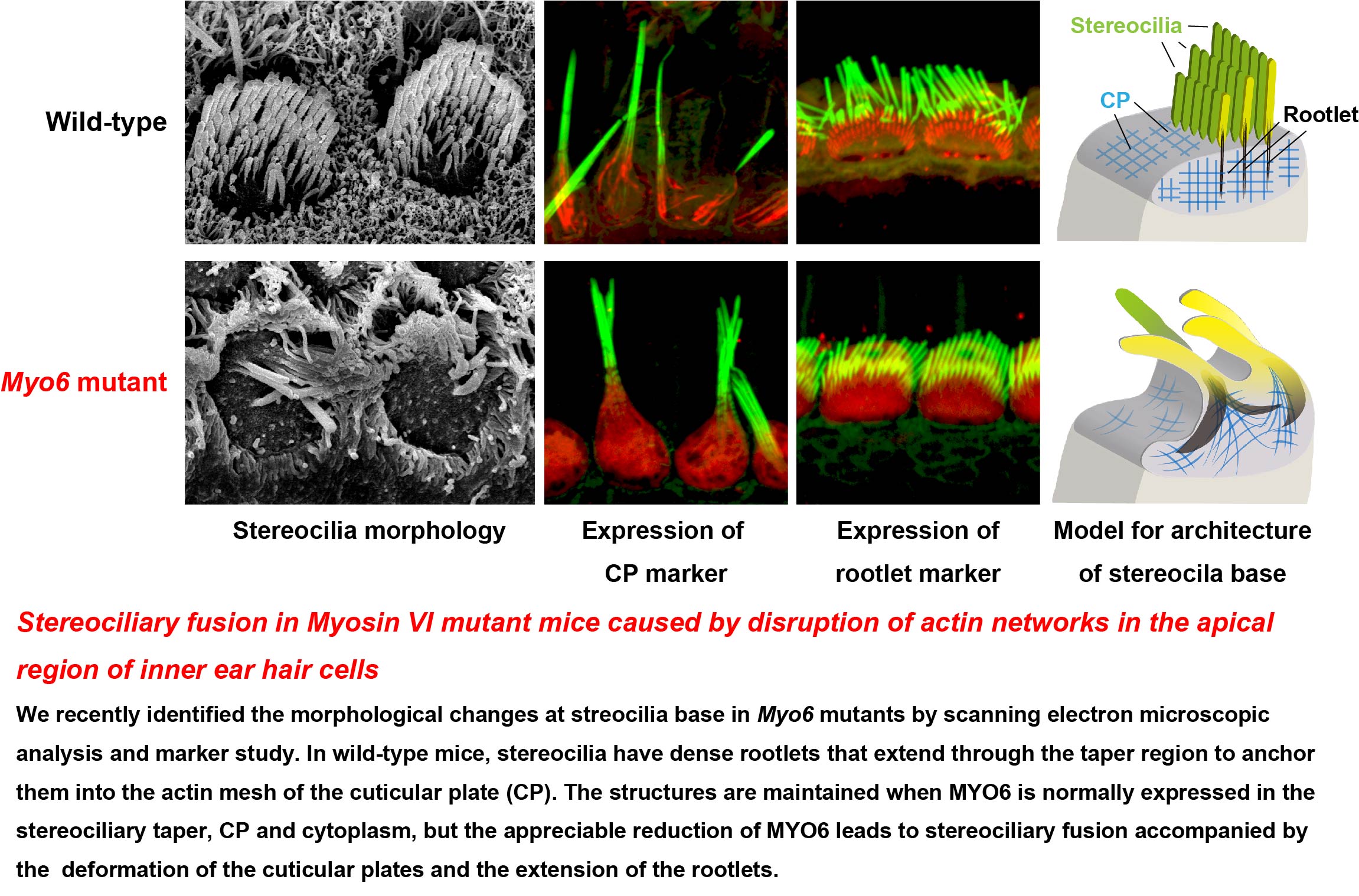
A novel splice site mutation of myosin VI in mice leads to stereociliary fusion caused by disruption of actin networks in the apical region of inner ear hair cells
An unconventional myosin encoded by the myosin VI gene (MYO6) contributes to hearing loss in humans. Homozygous mutations of MYO6 result in nonsyndromic profound congenital hearing loss, DFNB37. Kumamoto shaker/waltzer (ksv) mice harbor spontaneous mutations, and homozygous mutants exhibit congenital defects in balance and hearing caused by fusion of the stereocilia. We identified a Myo6c.1381G>A mutation that was found to be a p.E461K mutation leading to alternative splicing errors in Myo6 mRNA in ksv mutants. An analysis of the mRNA and protein expression in animals harboring this mutation suggested that most of the abnormal alternatively spliced isoforms of MYO6 are degraded in ksv mice. In the hair cells of ksv/ksv homozygotes, the MYO6 protein levels were significantly decreased in the cytoplasm, including in the cuticular plates. MYO6 and stereociliary taperspecific proteins were mislocalized along the entire length of the stereocilia of ksv/ksv mice, thus suggesting that MYO6 attached to taper-specific proteins at the stereociliary base. Histological analysis of the cochlear hair cells showed that the stereociliary fusion in the ksv/ksv mutants, developed through fusion between stereociliary bundles, raised cuticular plate membranes in the cochlear hair cells and resulted in incorporation of the bundles into the sheaths of the cuticular plates. Interestingly, the expression of the stereociliary rootlet-specific TRIO and F-actin binding protein (TRIOBP) was altered in ksv/ksv mice. The abnormal expression of TRIOBP suggested that the rootlets in the hair cells of ksv/ksv mice had excessive growth. Hence, these data indicated that decreased MYO6 levels in ksv/ksv mutants disrupt actin networks in the apical region of hair cells, thereby maintaining the normal structure of the cuticular plates and rootlets, and additionally provided a cellular basis for stereociliary fusion in Myo6 mutants.
This work was supported by JSPS KAKENHI for Scientific Research (B) (JP23300160), Challenging Exploratory Research (JP15K15625), and Grant-in-Aid for Young Scientists (B) (JP15K18393).
Seki et al. 2017 PLoS One 12, e0183477.
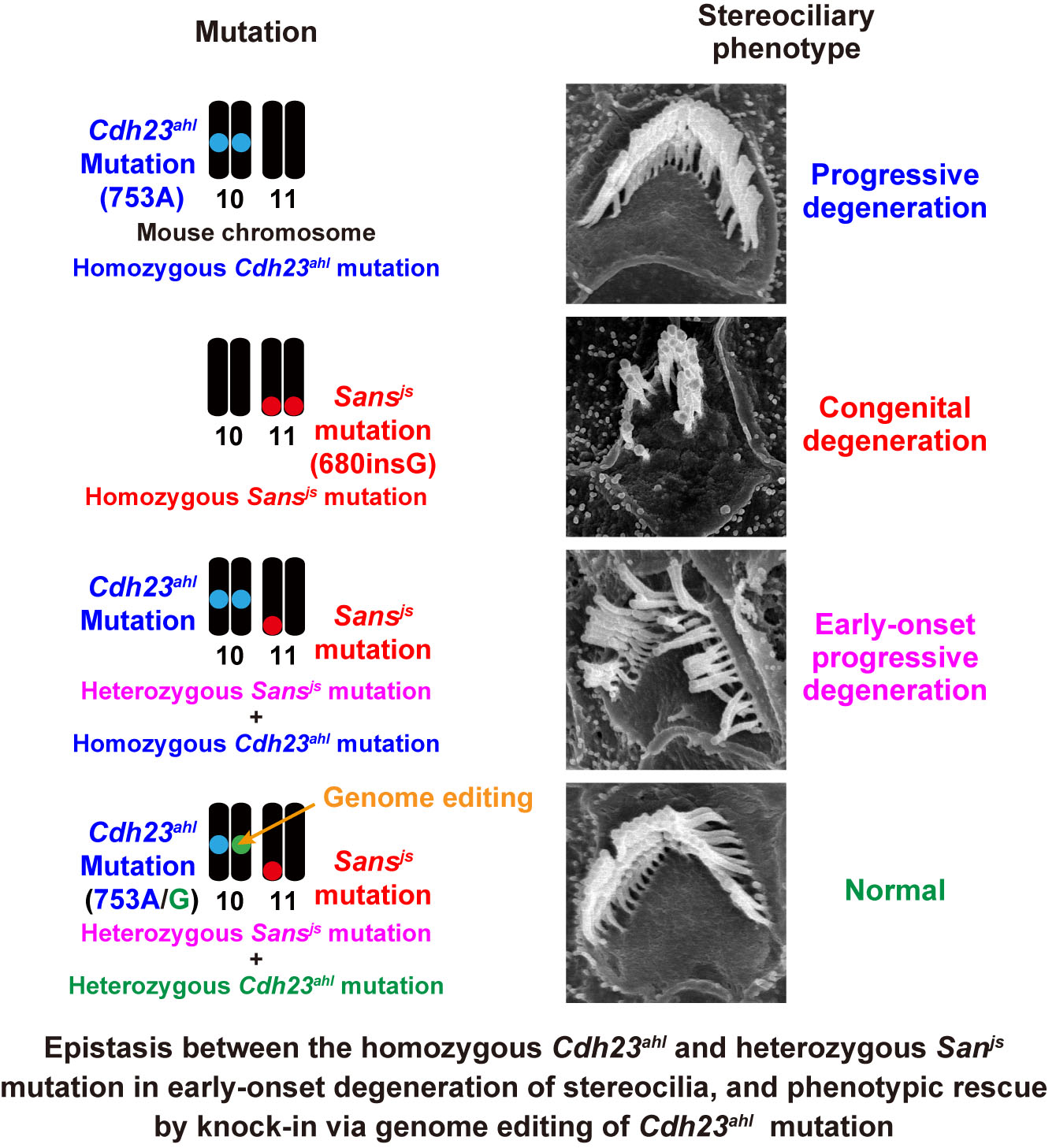
Heterozygous mutation of Ush1g/Sans in mice causes early-onset progressive hearing loss, which is recovered by reconstituting the strain-specific mutation in Cdh23
Most clinical reports have suggested that patients with congenital profound hearing loss have recessive mutations in deafness genes, whereas dominant alleles are associated with progressive hearing loss (PHL). Jackson shaker (Ush1gjs) is a mouse model of recessive deafness that exhibits congenital profound deafness caused by the homozygous mutation of Ush1g/Sans on chromosome 11. We found that C57BL/6J-Ush1gjs/+ heterozygous mice exhibited early-onset PHL (ePHL) accompanied by progressive degeneration of stereocilia in the cochlear outer hair cells. Interestingly, ePHL did not develop in mutant mice with the C3H/HeN background, thus suggesting that other genetic factors are required for ePHL development. Therefore, we performed classical genetic analyses and found that the occurrence of ePHL in Ush1gjs/+ mice was associated with an interval in chromosome 10 that contains the cadherin 23 gene (Cdh23), which is also responsible for human deafness. To confirm this mutation effect, we generated C57BL/6J-Ush1gjs/+, Cdh23c.753A/G double-heterozygous mice by using the CRISPR/Cas9-mediated Cdh23c.753A>G knock-in method. The Cdh23c.753A/G mice harbored a one-base substitution (A for G), and the homozygous A allele caused moderate hearing loss with aging. Analyses revealed the complete recovery of ePHL and stereocilia degeneration in C57BL/6J-Ush1gjs/+ mice. These results clearly show that the development of ePHL requires at least two mutant alleles of the Ush1g and Cdh23 genes. Our results also suggest that because the SANS and CDH23 proteins form a complex in the stereocilia, the interaction between these proteins may play key roles in the maintenance of stereocilia and the prevention of ePHL.
This work was supported by Japan Society for the Promotion of Science KAKENHI (Grants-in-Aid for Scientific Research B) (Grant Numbers 20300147, 23300160 and 15H04291 to Yoshiaki Kikkawa) and (Grants-in-Aid for JSPS Fellows) (Grant Number 14J06119 to Yuki Miyasaka).
Miyasaka et al. 2016 Hum Mol Genet 25, 2045-2059.
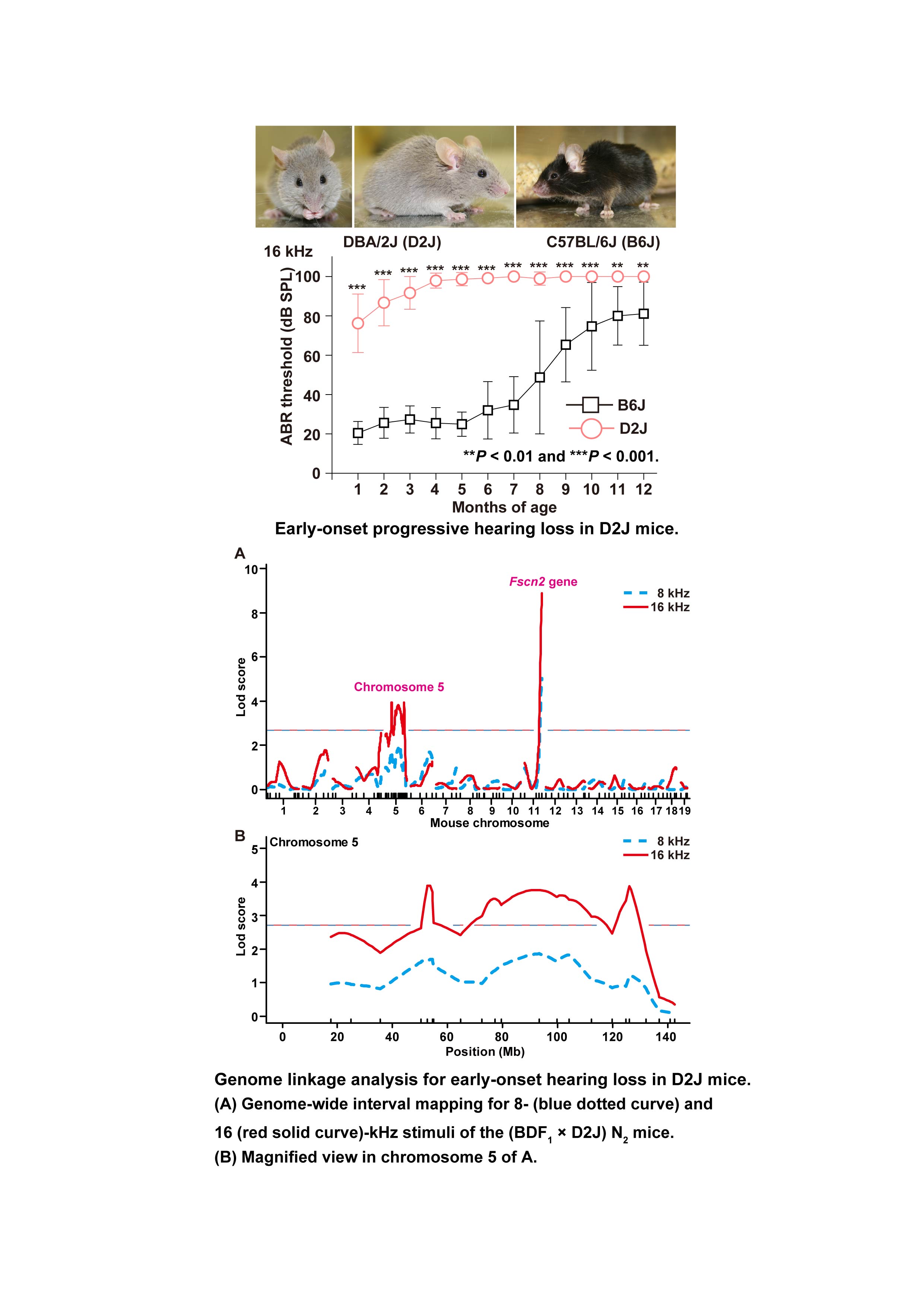
Quantitative trait loci on chromosome 5 for susceptibility to frequency-specific effects on hearing in DBA/2J mice
The DBA/2J strain is a model for early-onset, progressive hearing loss in humans, as confirmed in the present study. DBA/2J mice showed progression of hearing loss to low-frequency sounds from ultrasonic-frequency sounds and profound hearing loss at all frequencies before 7 months of age. It is known that the early-onset hearing loss of DBA/2J mice is caused by affects in the ahl (Cdh23ahl) and ahl8 (Fscn2ahl8) alleles of the cadherin 23 and fascin 2 genes, respectively. Although the strong contributions of the Fscn2ahl8 allele were detected in hearing loss at 8- and 16-kHz stimuli with LOD scores of 5.02 at 8 kHz and 8.84 at 16 kHz, hearing loss effects were also demonstrated for three new quantitative trait loci (QTLs) for the intervals of 50.3–54.5, 64.6–119.9, and 119.9–137.0 Mb, respectively, on chromosome 5, with significant LOD scores of 2.80–3.91 for specific high-frequency hearing loss at 16 kHz by quantitative trait loci linkage mapping using a (DBA/2J × C57BL/6J) F1 × DBA/2J backcross mice. Moreover, we showed that the contribution of Fscn2ahl8 to early-onset hearing loss with 32-kHz stimuli is extremely low and raised the possibility of effects from the Cdh23ahl allele and another dominant quantitative trait locus (loci) for hearing loss at this ultrasonic frequency. Therefore, our results suggested that frequency-specific QTLs control early-onset hearing loss in DBA/2J mice.
Suzuki et al. 2015 Exp Anim 64, 241-251.
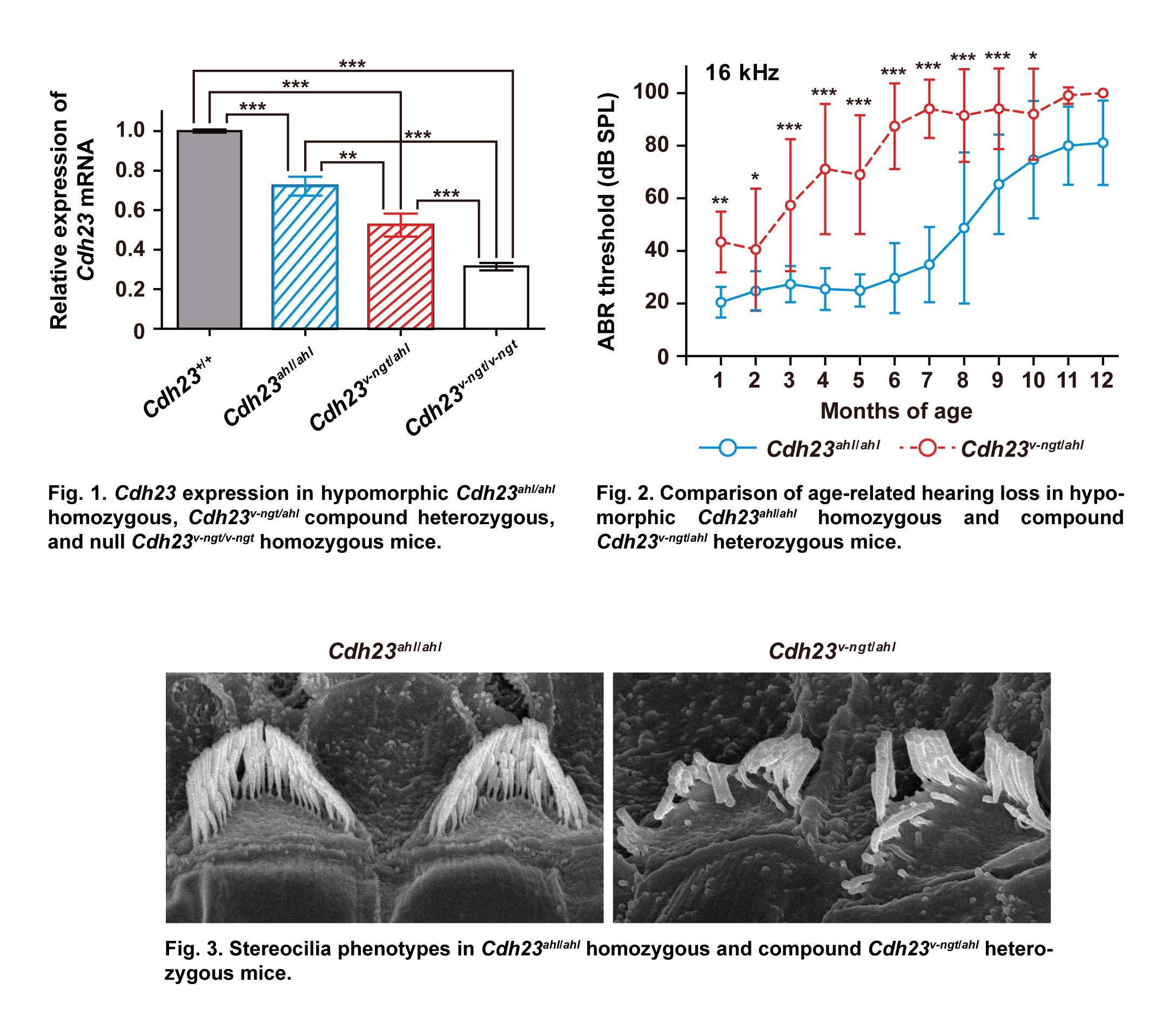
Compound Heterozygosity of the Functionally Null Cdh23v-ngt and Hypomorphic Cdh23ahl Alleles Leads to Early-onset Progressive Hearing Loss in Mice
The waltzer (v) mouse mutant harbors a mutation in Cadherin 23 (Cdh23) and is a model for Usher syndrome type 1D, which is characterized by congenital deafness, vestibular dysfunction, and prepubertal onset of progressive retinitis pigmentosa. In mice, functionally null Cdh23 mutations affect stereociliary morphogenesis and the polarity of both cochlear and vestibular hair cells. In contrast, the murine Cdh23ahl allele, which harbors a hypomorphic mutation, causes an increase in susceptibility to age-related hearing loss in many inbred strains. We produced congenic mice by crossing mice carrying the v niigata (Cdh23v-ngt) null allele with mice carrying the hypomorphic Cdh23ahl allele on the C57BL/6J background, and we then analyzed the animals' balance and hearing phenotypes. Although the Cdh23v-ngt/ahl compound heterozygous mice exhibited normal vestibular function, their hearing ability was abnormal: the mice exhibited higher thresholds of auditory brainstem response (ABR) and rapid age-dependent elevation of ABR thresholds compared with Cdh23ahl/ahl homozygous mice. We found that the stereocilia developed normally but were progressively disrupted in Cdh23v-ngt/ahl mice. In hair cells, CDH23 localizes to the tip links of stereocilia, which are thought to gate the mechanoelectrical transduction channels in hair cells. We hypothesize that the reduction of Cdh23 gene dosage in Cdh23v-ngt/ahl mice leads to the degeneration of stereocilia, which consequently reduces tip link tension. These findings indicate that CDH23 plays an important role in the maintenance of tip links during the aging process.
Miyasaka et al. 2013 Exp. Anim. 62, 333-346.
Advantages of a Mouse Model for Human Hearing Impairment
Hearing is a major factor in human quality of life. Mouse models are important tools for discovering the genes that are responsible for genetic hearing loss, and these models often allow the processes that regulate the onset of deafness in humans to be analyzed. Thus far, in the study of hearing and deafness, at least 400 mutants with hearing impairments have been identified in laboratory mouse populations. Analysis of through a combination of genetic, morphological, and physiological studies is revealing valuable insights into the ontogenesis, morphogenesis, and function of the mammalian ear. This review discusses the advantages of the mouse models of human hearing impairment and highlights the identification of the molecules required for stereocilia development in the inner ear hair cells by analysis of various mouse mutants.
Kikkawa et al. 2012 Exp Anim 61, 85-98.
