
Dr. Chihiro Hisatsune (Synaptic plasticity Project at TMIMS) et al. published an article entitled “Tuberous sclerosis complex (TSC) inactivation increases neuronal network activity by enhancing Ca2+ influx via L-type Ca2+ channels” in Journal of Neuroscience.
※ JNeurosci Featured Research
URL:https://www.jneurosci.org/content/featured-research ![]()
Twitter:https://twitter.com/SfNJournals/status/1438607638888132621 ![]()
Tuberous sclerosis complex (TSC) is an intractable disease characterized by benign tumors called hamartomas in the brain, lungs, and kidneys. The patient is known to develop neurological symptoms such as epilepsy, intellectual disability, and autism. Epilepsy, especially, is known to occur in as many as 80% of patients with TSC, and is highly associated with intellectual disability and quality of life of patients.
The causal genes for TSC are TSC1 and TSC2. They act as a GTPase activating protein for the small G-protein, Rheb. Loss-of-function mutations in either TSC1 or TSC2 increase the GTP-bound form of Rheb, resulting in the hyperactivation of the mammalian target of rapamycin complex 1 (mTORC1) and the subsequent neurological symptoms. However, the precise mechanism by which mTORC1 causes epilepsy is largely unknown.
The research group established human iPS cells lacking TSC2 using CRISPR-Cas9 system. They differentiated the TSC2-deficient iPSCs into cortical excitatory neurons and examined their physiological properties in culture. Using Ca2+ imaging, they first examined spontaneous activity of TSC2-deficient neurons (thereafter called as TSC2 neurons) and found that TSC2 neurons exhibit highly synchronized spontaneous neuronal activity with Ca2+ spikes. They also found TSC2 neurons have abnormal extended axons. Since both synchronized spontaneous neuronal activity and abnormal axon extensions in TSC2 neurons was suppressed by an mTOR inhibitor, it is likely that intimate neural networks with abnormally extended axons contribute to the synchronized neuronal network activity of TSC2 neurons.
The research group further examined Ca2+ signaling of individual neurons. They interestingly found that TSC2 neurons show enlarged Ca2+ influx upon membrane depolarization as compared to wild-type neurons (Figure 1). This enlarged Ca2+ influx was not observed in mTOR inhibitor-treated TSC2 neurons. Moreover, overexpression of constitutively active Rheb increases LTCCs expression and triggers enlarged Ca2+ influx upon membrane depolarization. Consequently, they concluded that elevated LTCC expression caused by mTOR activation cause enlarged Ca2+ influx in TSC2 neurons.
Finally, they examined the effect of enlarged Ca2+ influx on abnormal axonal extensions in TSC2 neurons. For this, they treated TSC2 neurons with an inhibitor for LTCCs and found that the LTCC inhibitor significantly suppressed axonal extensions of TSC2 neurons as compared to wild-type neurons. In addition, they also found that enhanced Ca2+ influx via LTCCs triggered sustained activation of CREB, a transcription factor that is important for synaptic plasticity. The sustained activation of CREB in TSC2 neurons upon depolarization became to be transient like in wild-type neurons, when TSC2 neurons were treated with mTOR inhibitor.
This study showed that TSC-mTOR signaling regulates LTCC expression in neurons. In addition, the study showed that increased Ca2+ influx via LTCCs in TSC2 neurons contributed to the abnormally extended axonal elongation and sustained activation of the transcription factor, CREB (Figure 2). It is well known that intracellular Ca2+ concentration level has an intimate relationship with neuronal activity. The results of this study suggest an important connection between sustained mTOR activation and elevated Ca2+ signaling via LTCC in human TSC neurons, which could cause epilepsy in TSC. It is important for future studies to examine whether the neurons of patients with TSC have enhanced Ca2+ influx via LTCCs and altered LTCC levels in the brain.
This work was supported by a Grant-in-Aid for Scientific Research (B), Grant Number JP25293239/18H02536 (KanatoYamagata), the Japan Agency for Medical Research and Development (grant number JP18ek0109311h0001/19ek0109311h0002/ 20ek0109311h0003, (KanatoYamagata), the SENSHIN Medical Research Foundation (KanatoYamagata), a National Institutes of Health Grant (R01EY026817, Amy Lee), and a Grant-in-Aid for Scientific Research (C), grant number 19K08334 (Chihiro Hisatsune).
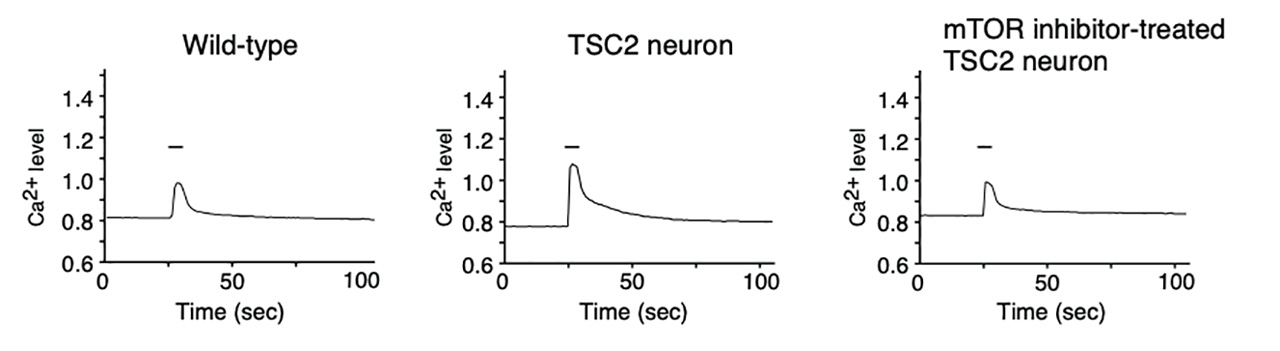
Figure 1. TSC2 neurons showed enhanced Ca2+ influx upon membrane depolarization
Ca2+ response upon membrane depolarization in wild-type (left) and TSC2 (middle) neurons. mTOR inhibitor treatment ameliorates enhanced Ca2+ influx in TSC2 neurons (right). Bars represent 60 mM KCl stimulation for 20 sec.
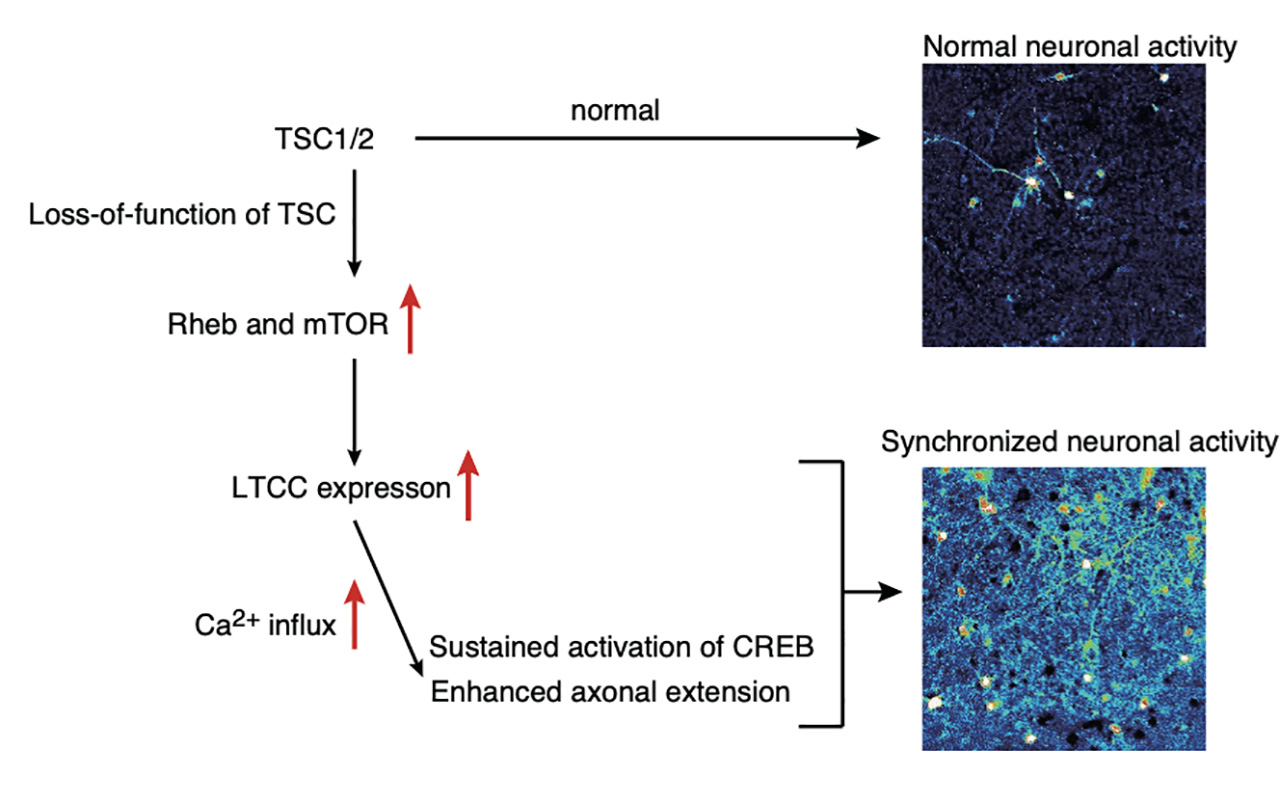
Figure 2. A schematic model of the mechanism of enhanced activation of TSC neurons
In normal neurons, only parts of the neurons are activated, spontaneously. Loss of function of mutations in TSC1 or TSC2 increases Rheb, resulting in hyperactivation of mTOR. Hyper-activated mTOR increases LTCC expression and cause large Ca2+ influx upon neuronal activation in TSC neurons. Enlarged Ca2+ influx cause abnormal axonal extensions and sustained activation of CREB. These physiological changes may trigger epilepsy in TSC.
