医学・生命科学全般に関する最新情報





![]()


![]()
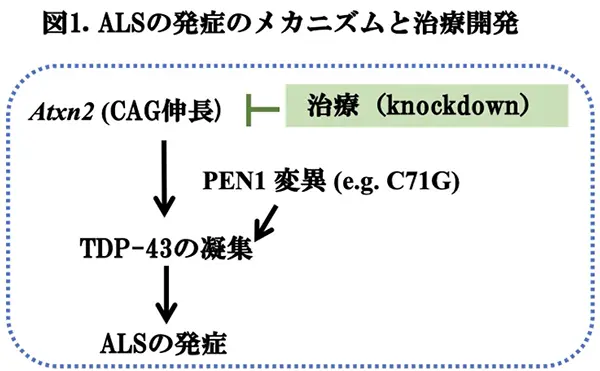
ALSは、上位運動ニューロンと下位運動ニューロンの両者の細胞体が散発性・進行性に変性脱落する神経変性疾患です。ALSの病態における神経毒性の中心は、TDP-43の凝集ですから、この病理的メカニズムを理解することが治療開発に重要です。TDP-43以外にも、多くの蛋白凝集がALSの病態に関与することが知られていますが、中でも、Ataxin-2とプロフィリン-1 (PFN1)は、TDP-43の凝集促進のリスク因子として重要であると考えられて来ました(図1)。Ataxin-2をコードするAtxn2は、12番染色体長腕(12q24.12)に座位する遺伝子であり、その翻訳領域における通常はアミノ酸残基(a.a.)23以下のCAGリピートが異常伸長した(>35〜40 a.a.)ポリグルタミン鎖を持つタンパク質(PolyQ)が生成されます。この異常PolyQは、神経細胞内で凝集・蓄積することにより、神経変性を引き起こし、脊髄小脳変性症2型*4が発症します。興味深いことに、Atxn2のCAGリピートの中間的伸長(27~34 a.a.)は、ALSの病態にリンクすることが知られており、TDP-43の凝集促進に関与するかも知れないと考えられています(図1)。一方、アクチン結合タンパク質の一種であるPFN1は遺伝子変異により凝集性を獲得し、TDP-43の凝集をする可能性が示されてきました。今回、Ataxin-2がALSの治療標的になる可能性を検討するために、米国Biogen社のZachary C. E. Hawley博士らは、変異型PFN1C17Gを過剰発現したALSのTgモデルマウス(内因性TDP-43の凝集・蓄積)を作成し、このマウスに対して、アデノ随伴ウイルス(AAV)を用いたmiRNAによる遺伝子治療を行ったところ、中枢神経系(CNS)におけるTDP-43の凝集・蓄積は抑制され、運動機能は改善することを見出し、ALSの治療開発への応用が示唆されました。研究成果は、最近のActa Neuropathol Commun.誌に掲載されましたので、この論文(文献1)を報告いたします。ALSにおいては、抗TDP-43モノクローナル抗体を用いた免疫療法などの治療開発が進んでいないことを考慮しますと、これらの結果は非常に魅力的です。
文献1.
Hawley ZCE, et al, Viral-mediated knockdown of Atxn2 attenuates TDP-43 pathology and muscle dysfunction in the PFN1C71G ALS mouse model. Acta Neuropathol Commun 2025 13(1):116.
ALSは、進行性運動ニューロンの喪失と筋萎縮を特徴とする致死性の神経変性疾患であり、CNSにおけるRNA結合タンパク質TDP-43の高リン酸化凝集は、TDP-43の機能不全がALSの病態形成の根底にあることを示唆している。これをTgマウスモデルにおいて検討することを研究の目的とする。
本研究では、ALS関連PFN1C71G変異タンパク質を発現するTgマウスを作成し、これをモデルにしてAtxn2のノックダウンによる治療効果を検討する。